1. Introduction
Atomic layer deposition (ALD) is one of the most attractive methods to grow uniform, conformal thin films with a high degree of control over film thickness and composition due to its self-limiting surface reactions.1) In ALD the film grows through a cycling of self-limiting surface reactions. Ideally, each exposure of the surface to the gas-phase precursor results in, at most, one monolayer of growth via a self-terminating reaction. For example, in the ALD of a binary oxides, the growth cycle consists of exposure of the surface substrate to a metal precursor followed by exposure to an oxygen precursor, separated by a purge period. For metallization, ligand loss and elemental reduction is achieved by alternating exposure to gas-phase reducing agents. Consequently, ALD affords uniform and conformal deposition of a material, with thickness control at the atomic layer level.
The general ALD half-reaction mechanism is adsorption of the precursor on the surface, followed by ligand exchange/release, and finally desorption of product; with the ALD productive reaction pathway in competition with other potential reactions available to the system. As with any chemical reaction, the rate of reaction, mechanistic path, and chemo-/regio-selectivities are directly determined by the free energies of the critical point structures defining a particular reaction pathway on the ALD substrate. The number, diversity and complexity of the surface-precursor reactions reflect the complexity and heterogeneity of the precursor metal-ligand architecture. ALD precursor ligands vary greatly in complexity from halides, to alkyls and heteroalkyls, to bi- and higher-dentate coordinate ligands; in a homoleptic or heteroleptic coordination environment. On the other side of the process, ALD co-reactants have the potential for similar or greater chemical diversity. This chemical diversity provides great opportunity for chemical design to achieve the diverse physical properties and enhanced reactivity needed for thin film deposition in the fabrication of next-generation electronic devices. However there is a pressing need for the development of efficient and low-cost methods to systematically explore ALD precursors chemical design space and advise experimental efforts.
Over the past two decades, advances in quantum mechanics simulation codes based on density functional theory (DFT), and the rapid improvement in the performance of computational resources have dramatically increased the applicability of first-principles modeling from the small molecule domain to increasingly larger and more chemically realistic systems; with higher and higher accuracy. Homogeneous catalysis is an industrial application that has many parallel concerns to ALD precursor development. DFT has been shown to be an invaluable tool for furnishing atomistic details of reaction mechanisms, reliably predicting reaction energetics with accuracy comparable to experimental kinetic measurements; allowing the rational modification of single-site catalysts to achieve desired increases in reactivity and chemo-, regio-, and stereo-selectivity.2-4) Similarly, DFT is an invaluable tool applied to ALD precursor reactivity and performance; improving the understanding of structure- property relationships, providing new details about growth and failure pathways. A significant number of reports have been made applying DFT to investigate ALD processes (see review by Elliott5)), however it has not been as widely adopted as in the catalysis community. Even more compelling is the potential for the discovery of new ALD precursors and processes accelerated by DFT simulation. Key to the realization of this exciting next step, is to move predictive chemical simulation from the traditional explanation or analysis role, to the front of the ALD precursor development process to filter precursor design space, using automated simulation and virtual screening approaches.
In the present work, a perspective is given on the application of quantum mechanics simulation (e.g. DFT) to ALD precursor and process analysis, and discovery. The application of DFT to ALD reactivity is illustrated by analysis of the reaction of the archetype ALD precursors for deposition of Al2O3, trimethylaluminum (Al(CH3)3, TMA) and H2O, with a hydrogen-terminated silicon substrate (H/Si(111). The application of virtual screening for ALD precursor modification and optimization is illustrated for two cases: the thermochemistry of Al-precursor nucleation (heterodeposition) on H/Si, and the kinetics and thermochemistry of Siprecursors for Si3N4 growth (homodeposition).
2. Methods
The results presented here were obtained using the Schrödinger Materials Science Suite (Version 16-1).6) All DFT calculations used the Jaguar electronic structure program (Version 9.1).7,8) The minimum energy structures and energies for reactants, surface complexes, transition structures and products (as described in text) were computed using the M06- L local density functional,9) and the B3LYP hybrid density functional,10,11) along with the standard double-ζ polarized Pople basis set, 6-31G**.12) The nature of the optimized structures were confirmed by subsequent harmonic frequency calculations, which provides temperature dependent free energies. The effect of chemical substitution on relative energies for example ALD reactions was calculated using the Reaction Energetics Enumeration module which automatically enumerates diverse structure libraries (R-group based scheme) for reaction related structures and launches the required DFT simulations to evaluate energy differences.
3. Quantum Mechanical Analysis for ALD Precursor Reactivity and Processes
There is pressing need in the semiconductor industry for the rapid development of metal precursors and functional co-reactants that deliver the required surface reactivity and selectivity for next-generation thin-film deposition processes. DFT simulation can provide critical insight into the fundamental properties, energetics and atomistic details of the key reaction pathways that are central to the ALD process; often furnishing details that are too costly or not easily accessible for measurement using experimental techniques. Typical properties that can be calculated for ALD precursors, adsorption complexes, transition states and surface half-reaction products include: minimum energy conformations and geometries, relative energies (e.g. Gibbs free energies (ΔGr, ΔG‡), determining kinetic and thermodynamic favorability), and characteristic spectroscopic signatures (e.g. infrared bands and associated normal modes, useful in assisting experimental monitoring of the surface structure and evolution during an ALD process). For a given reactive precursor interacting with an ALD substrate, there are multiple reaction pathways that are possible, depending on the structure and chemical composition of the precursor and surface. DFT simulations analyzing the relative energies of competing pathways can provide a rationalization of the observed ALD reaction rates and products. Moreover, the effect of changes in chemical structure of the precursor, or termination of the ALD substrate, can be evaluated in attempt to tailor the chemistry to improve the energetics and specificity of the ALD process.
The most widely studied ALD process both experimentally 13) and computationally14,15) is the deposition of Al2O3 using trimethylaluminum (TMA, Al(CH3)3) and H2O. TMA is the preferred aluminum precursor for ALD of Al2O3 due to its thermal stability, high vapor pressure (8.4 Torr) and highly exothermic reaction with H2O. Growth of Al2O3 by ALD using TMA and H2O is generally regarded as the model ALD process; with nearly ideal ligand exchange and selflimiting behavior. The TMA and H2O ALD growth half-reactions (R1 and R2, respectively) can be denoted as:
where asterisks denote surface species. Subsequent alternating exposures of the hydroxylated alumina surface to TMA and H2O, leads to formation of Al-O linkages with loss of CH4, giving growth of Al2O3. In ALD, there are at least two different deposition stages: growth (homodeposition) as described above in R1/R2 for Al2O3 ALD, and nucleation (heterodeposition) involving the formation of the interface layer on a chemically distinct surface substrate. The optimization of an ALD process requires careful consideration of the efficiency and kinetics of both deposition stages. Due to the need for atomically abrupt oxide interfaces, minimizing formation of a deleterious SiO2 interface layer, there is interest in developing ALD processes on hydrogen-terminated silicon (H/Si). Precursors that perform excellent for ALD growth, can have dramatically lower reactivity in the nucleation stage of deposition. Additionally, effective ALD nucleation depends on the order of precursor exposure. In the case of Al2O3 deposition on H/Si, Frank, Chabal and Wilk carried out a critical spectroscopic study characterizing the process and products of interface formation using TMA and H2O.16) In that work they report that, “Contrary to common belief, we find that the metal precursor, not the oxidizing agent, is the key factor to control Al2O3 nucleation on hydrogen-terminated silicon”. Under the conditions of the ALD experiment, TMA was shown to react with H/Si preferentially over H2O at 300°C. Here to illustrate the predictive capability of DFT applied to ALD processes and precursor chemistry, calculations are presented extending and confirming previous work17,18) analyzing the chemical pathways and controlling energetics for TMA and H2O interacting with H/Si.
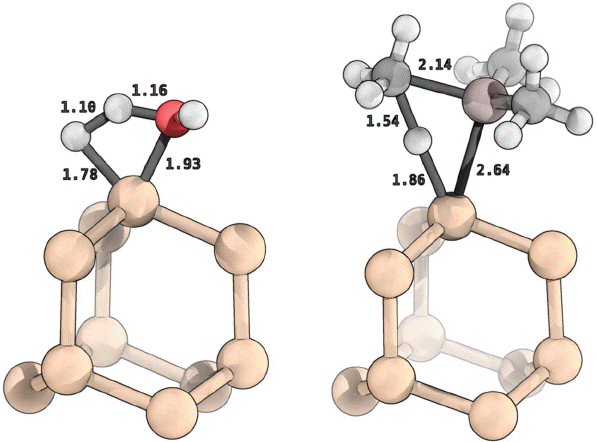
In this work, a discrete cluster model is used to represent the ALD reaction site on the silicon surface. The surface reaction site model corresponds to a hydrogen-terminated Si cluster with stoichiometry Si10H16, representing roughly one unit cell of the H/Si(111) surface. Minimum energy structures, vibrational frequencies and free energies were calculated for the reactants, transition structures and product species for the dominant reactions of TMA and H2O with the H/Si(111) model using the M06-L/6-31G** level of theory. The TMA and H2O ALD nucleation reactions investigated here (R3 and R4, respectively) can be denoted as:
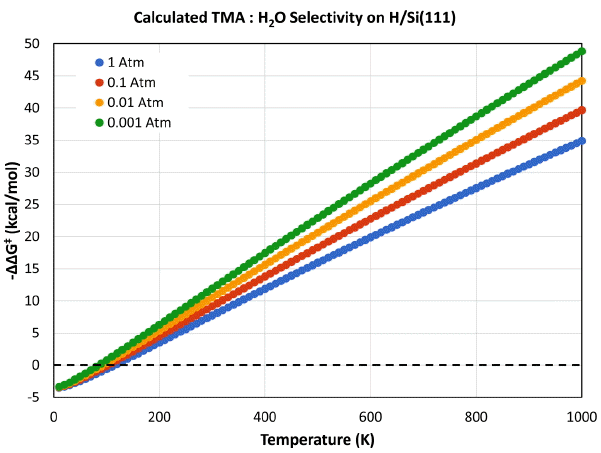
where asterisks denote surface species. The initial reaction of the hydrogen-terminated Si surface with TMA or H2O, can lead to formation of Si-Al or Si-O linkages, with release of CH4 or H2 respectively. The reaction free energy profiles at 300 K (1 atm) for the surface TMA and H2O reactions are shown in Fig. 1 (I, II, III critical points correspond to gasphase precursor, surface transition state and product; with H2O energies and TMA energies colored red and purple, respectively). As presented there, the two precursor nucleation reactions differ in terms of relative kinetic and thermodynamic favorability. The H2O + H/Si reaction is calculated to be more thermodynamically favored with a ΔGR of -32.1 kcal/mol, whereas the TMA + H/Si product is less exergonic with a ΔGr of −0.5 kcal/mol. The transition structures for the reactions are shown in Fig. 2; with the transitional bond lengths reported. The transition state relative free energies controls the kinetics of reaction, with the rate being an exponential function of the free energy barrier, ΔG‡. Shown in Fig. 1, the relative energies for the transition structures clearly resolve the two reactions with the ΔG‡ for the TMA reaction being lower than the H2O reaction by 7.7 kcal/mol (40.2 compared to 47.9 kcal/mol). Using the computed activation energies and thermodynamic functions for the TMA or H2O + H/Si reactions, the nucleation reaction selectivity can be estimated for different reaction conditions. The kinetic barrier difference (ΔΔG‡) at 200, 300 and 400 K is 3.5, 7.7 and 11.0 kcal/mol, which gives reaction ratios of 370:1, 400,000:1 and 450,000,000:1 in favor of TMA, respectively. Fig. 3 shows the H/Si Al2O3 nucleation reaction TMA:H2O selectivity as a function of temperature with varying pressure (1, 0.1, 0.01 and 0.001 atm). The results presented in Fig. 3, show that TMA is more reactive than H2O with H/Si at conditions with pressure > 0.001 atm and temperature > 80 K. The computational analysis clearly predicts that for nucleation of Al2O3 ALD on H/Si requires reaction with TMA, not H2O, in excellent agreement with experimental observations.
As shown for Al2O3 ALD nucleation using TMA and H2O, DFT is an invaluable tool for predicting the energetics controlling ALD reactivity and selectivity; having the potential to provide a priori guidance for precursor selection, and process optimization.
4. Simulated Spectroscopy for ALD
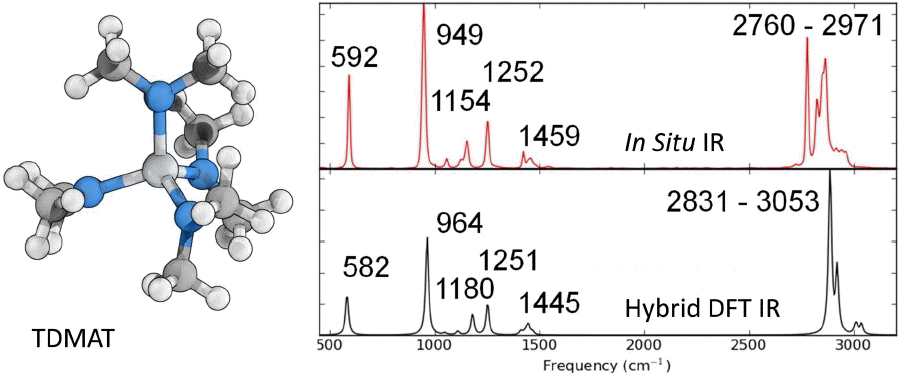
In situ vibrational spectroscopy has been shown to be a powerful tool for ALD structure characterization and process monitoring.19-22) Infrared (IR) absorption spectroscopy allows the determination of the local chemical structure of the active ALD surface allowing elucidation and confirmation of chemical mechanism and reaction products; provided a reliable correlation can be made between experimentally observed bands and the constituent chemical motifs. Interpretation of IR for ALD structures is challenging because of loss of symmetry upon precursor adsorption, bonds broken/formed through reaction, and limitations in measurement at low concentration (monolayer coverage) and strong polarization effects. DFT can help provide conclusive IR band assignments, allowing reliable interpretation of experimental spectra for ALD. Prediction of harmonic frequencies, spectral intensities, and normal modes allows construction of a simulated IR spectrum for comparison with experimental data to aid assignment. To illustrate the predictive capability of DFT for IR spectra related to ALD, Fig. 4 shows the experimentally measured in situ IR spectrum for the titanium precursor, tetrakis(dimethylamino)titanium (TDMAT),23) along with the hybrid DFT computed IR spectra for comparison. To correct for the neglect of anharmonicity, a homogeneous scaling factor of 0.9725 is applied.24) This leads to better correlation between computed and measures frequencies, however individual vibrational modes may be slightly overestimated or underestimated depending on the details of the particular mode. As shown in Fig. 4, the agreement between the calculated and measured IR bands is excellent, both qualitatively and quantitatively. For TDMAT, the Ti-N stretch and N-C stretch are observed at 592 and 949 cm−1, and are calculated at 582 and 964 cm−1. Similar agreement can be expected for surface structures in the ALD process. DFT simulated spectra for intact precursors, surface complex and ALD reaction products can provide conclusive experimental band assignments, which gives direct confirmation of the structures and reaction details for nucleation and growth processes in ALD.
5. Automated DFT Simulation for ALD Precursor Discovery
Interactive DFT simulation can be used to analyze structures, properties, reaction mechanisms as well as controlling energetics for known ALD precursors; rationalizing observed property and performance trends. More compelling is using the predictive capability for in silico precursor optimization and design. DFT can be used to explore the effect of changing precursor and/or surface chemical structure and composition and assessing novel molecular architectures to help prioritize precursor candidates that have the highest probability for delivering the desired thermal stability, ALD reactivity and selectivity.
The virtual screening paradigm in materials science is a recent possibility due to advances in computational power and the efficiency and stability of quantum mechanics simulation packages. Electronic structure codes are extremely robust for standard types of analyses, usually requiring no user intervention once the physical system and parameters have been set and the calculation initiated. This makes it possible for individual DFT calculation steps to be executed in sequence comprising an automated simulation workflow, in which candidate precursors of varying structure and composition are analyzed in an automated fashion with the results collected in a growing data record. This record can then be sorted and mined to identify lead ALD precursors candidates and establish critical structure-property limits within a given chemical design space. This automated DFT simulation approach is illustrated to explore the chemical design space of Al precursors, assessed by the enthalpy of reaction for the nucleation reaction H/Si(111) computed using B3LYP/6-31G**.
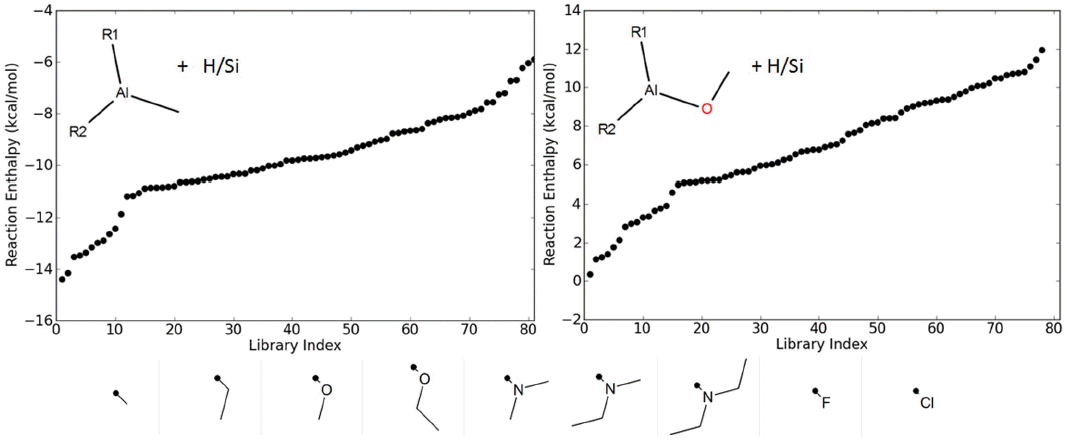
The Al-precursor chemical design space screened in this work is defined as R1R2Al-X, where X is the leaving group, either -CH3 or -OCH3, and R1, R2 are independently selected from the nine electron withdrawing groups (EWG) and electron donating group (EDG) substituents shown in Fig. 5 (lower). This chemistry produces 81 precursor candidates per leaving group. The reaction of interest is the analogous reaction to R4, with the substituted Al-precursor reacting with the H/Si surface forming a Al-Si bond, with loss of HX (CH3 or HOCH3). The reaction site is modeled by a Si10H16 cluster, as in Section 3.
The overall internal enthalpy for R1R2Al-X reacting with H/Si is calculated using an automated DFT reaction energy module at the B3LYP/6-31G** level of theory (from reaction complex to product complex) for the -CH3 and -OCH3 precursor libraries. Fig. 5, left and right, summarizes the calculated H/Si nucleation reaction enthalpies (0 K) for the Al-CH3, and Al-OCH3 precursor libraries, respectively. Comparison of the reaction energetics clearly shows the difference in leaving group, with the all -CH3 precursor H/Si reactions being exothermic, whereas the -OCH3 reactions are endothermic. Looking at the range of enthalpies gives indication of how much the thermodynamic favorability for the -CH3 and -OCH3 leaving group reactions can be tuned by EDG or EWG substitution. For the methane formation library screen, the range of reaction enthalpies is ca. 8.5 kcal/mol (−14.4 to -5.91 kcal/mol), and the methanol formation library enthalpies span from nearly thermoneutral at 0.33 kcal/mol to being endothermic by 11.9 kcal/mol. Interestingly substitution of the EDG or EWG has the inverse effect on R1R2Al-CH3 and R1R2Al-OCH3 enthalpies; EDG or EWG substitution increasing or reducing the thermodynamic favorability for Al-Si and CH4 formation, respectively, with the opposite trend predicted for Al-Si and HOCH3 formation.
Next, another simple example illustrating the use of automated simulation to explore ALD precursor design space is discussed for Si-containing precursors selected for ALD of silicon nitride. Low temperature Si3N4 is a challenging thermal ALD process. Si3N4 ALD using dichlorosilane,25,26) silicon tetrachloride,27,28) and silicon hexachloride29,30) as Si precursors along with NH3 have been experimentally reported. In this work, automated DFT calculations are used to assess the reaction enthalpy and kinetic barriers for a variety of Si containing precursors through a gas-phase reaction representative of the surface ALD half-reaction. The scope of the analysis include examination of several well-known precursor compounds: chlorosilane, tetrachlorosilane and dichlorosilane, as well as a number of commercially available precursors with methyl groups such as SiCl2(CH3)2 (by Gelest) and Cl3SiCH3 (by Sigma Aldrich).
In total, 19 silicon precursors were analyzed defined by the following substitution schemes: SiClR3 with R= H, Cl, or CH3, and Si2ClR5 with R= H or Cl. The Si precursor growth half-reactions that were considered can be denoted as:
where asterisks denote surface species. For the automated DFT screening of the candidate Si precursors, the simplest model was used representing the NH-terminated Si3N4 surface by NH3. This simple model should be useful to assess electronic effects across the library, though steric interactions may not be adequately represented. However, calibration of the predicted energetics for the simple NH3 reactions against results obtained with a larger Si3N4 cluster model showed that the NH3 reaction calculations reproduce the kinetics barriers within 3 kcal/mol.
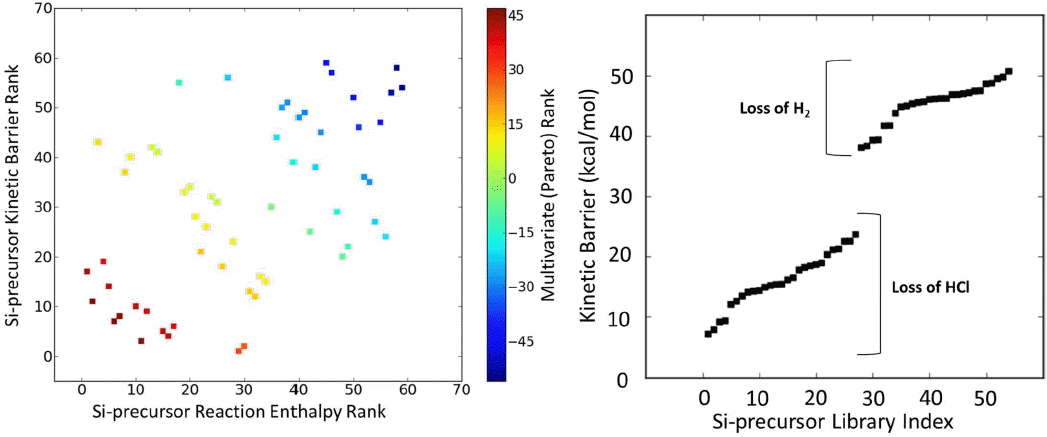
An optimal precursor for an ALD process must simultaneously satisfy a number of target objectives related to ALD process performance; such as stability, kinetics, thermodynamics, and selectivity. Multi-objective solutions represent a trade-off between objectives, with one class being Paretooptimal solutions. Pareto-optimal solutions are defined as a set of solutions which are non-dominated, such that it is not possible to improve one property without making any other property worse.31) The Si precursor library was Pareto rank ordered based on the computed reaction enthalpies and kinetic barriers, looking to minimize the kinetic barrier and maximize the thermodynamic favorability. A scatter plot of ranking for Si precursor enthalpy and barrier is shown in Fig. 6 (left); with the candidate structure data points colored by their Pareto ranking. Optimal structures correspond to those colored dark red, plotted in the lower left quadrant. Among the highest ranked precursors were Si(CH3)Cl3 and Si2HCl5. Fig. 6 (right) shows an ordered plot of the B3LYP/6-31G** calculated barriers for the model Si3N4 ALD reaction. Consideration of the barriers across the library shows that the reactions resolve themselves into two groups depending on where the reaction formed H2 or HCl. The results indicate the removal of precursor reaction with loss of H is more kinetically hindered than the loss of Cl. This analysis reveals that the known precursors for Si3N4 deposition, such as chlorinated silanes do show low energy barrier with relatively favorable thermodynamics. In addition, several new candidates were identified that have not been previously studied. These molecules have favorable reaction energetics and show good potential as low-T Si3N4 ALD precursors.
In this work, illustrative examples of interactive and automated quantum mechanical simulation approaches for ALD precursor reactivity and discovery were presented. Beyond the conventional use of DFT analysis to investigate and understand the structure, controlling energetics and reactivity of existing ALD precursors, greater impact is expected for in silico evaluation of new classes of potentially disruptive reactive precursors with enhanced and differentiated reactivity. Currently, in close collaboration with experimental efforts, simulation assisted discovery is being applied to accelerate discovery and development of new ALD molecule to material processes to meet the requirements for next generation microelectronic devices.
6. Outlook and Conclusions
Quantum mechanical simulation is an extremely powerful tool applied to ALD precursor reactivity and performance; providing reliable details about structure and reaction mechanisms, and the controlling energetics determining the thermodynamic and kinetic favorability of ALD nucleation and growth pathways. The predictive capability was illustrated for the case of Al2O3 ALD nucleation on hydrogen-terminated silicon (H/Si). The use of automated DFT simulations to estimate structure-reactivity ranges and identify lead candidates within a given chemical design was demonstrated for two simple cases, thermodynamics of Al-precursor nucleation for Al2O3 ALD on H/Si, and thermodynamic and kinetic screening of Si-precursors for Si3N4 ALD. The predictive capability of DFT coupled with robust automation extends the role that atomic-scale simulation can play from one of explanation and elucidation after experimental realization, to one of optimization and discovery of new metal-ligand architectures and functional co-reactants with enhanced reactivity and selectivity, informing synthetic ALD precursor development and driving innovation in the development of next-generation ALD precursors and processes.