Investigation of LiO2 Adsorption on LaB1−xB′xO3(001) for Li-Air Battery Applications: A Density Functional Theory Study
Article information
Abstract
Li-air batteries have received much attention due to their superior theoretical energy density. However, their sluggish kinetics on the cathode side is considered the main barrier to high performance. The rational design of electrode catalysts with high activity is therefore an important challenge. To solve this issue, we performed density functional theory (DFT) calculations to analyze the adsorption behavior of the LiO2 molecule, which is considered to be a key intermediate in both the Li-oxygen reduction reaction (ORR) and the evolution reaction (OER). Specifically, to use the activity descriptor approach, the LiO2 adsorption energy, which has previously been demonstrated to be a reliable descriptor of the cathode reaction in Li-air batteries, was calculated on LaB1−xB′xO3(001) (B, B′ = Mn, Fe, Co, and Ni, x = 0.0, 0.5). Our fast screening results showed that LaMnO3, LaMn0.5Fe0.5O3, or LaFeO3 would be good candidate catalysts. We believe that our results will provide a way to more efficiently develop new cathode materials for Li-air batteries.
1. Introduction
The use of fossil fuels has exacerbated various environmental concerns, particularly global warming. To reduce the dependence on fossil fuels, nowadays Li-air batteries, due to their high theoretical specific energy, have attracted interest for future electric vehicles, electrical energy storage devices, and other energy applications.1–3) However, many scientific issues such as low round-trip efficiency, short cycle life, poor rate capability, and electrolyte instability must be solved for the realization of affordable Li-air battery technology.4–9) Especially, the slow kinetic rate of the oxygen reduction reaction (ORR) or the oxygen evolution reaction (OER) on the cathode side has been widely known as a main obstacle to high energy efficiency.10) To overcome this problem, numerous attempts have been made to design new electrocatalysts with high activity for ORR or OER.11) In the past, perovskite oxides (ABO3) have been nominated as potential ORR or OER catalysts12,13) due to their high catalytic activity, fast electronic and ionic conductivity, and low cost.12,14,15) For these reasons, many researchers have made efforts to enhance the cathode kinetics using LaBO3-type perovskite oxides such as LaSrMnO3,16,17) LaFeO3,18) and LaSrCoO3−δ 19) in Li-air batteries. However, in spite of these constant efforts, it is still difficult to find optimal combinations of components in perovskite oxides due to changes in catalytic properties with partial or full substitution at A- and B-sites.
Previous studies have provided efficient ways to improve the catalytic properties. Introducing an activity descriptor enables us to facilitate fast screening of perovskite catalysts for Li-air batteries.14,15) Using density functional theory (DFT) calculations, Nørskov et al.20) suggested a method that can assess the thermochemistry of the electrochemical reactions. They also verified the origin of the overpotential for oxygen reduction over Pt(111). Based on these results, Man et al.21) derived a descriptor (ΔGO*–ΔGHO*) for oxygen reduction using different kinds of oxide materials. These results has been widely applied in various electrochemical reactions including Li-ORR and OER to introduce useful descriptors. Choi et al.22) used the DFT calculated adsorption energy database of lithium oxide intermediates to examine the theoretical overpotentials for ORR and OER on Pd, Cu, and PdCu alloy surfaces with an ordered body centered cubic (B2-type) structure. They reported that the LiO2 adsorption energy shows a good correlation with the ORR and/or OER overpotentials. Moreover, they demonstrated that controlling the LiO2 adsorption energy by alloying the material with Pd and Cu can enhance the catalytic activity. A similar argument was also made by Kim et al.,23) who suggested that the overpotential of Li-OER decreases with decreasing of the strength of LiO2 adsorption on Pt(111), Co(0001), and Pt3Co(111). These results motivated us to estimate the catalytic activity on perovskite oxides by using only a simple descriptor, the LiO2 adsorption energy.
In this study, we first use DFT calculations to perform a structural investigation of the LiO2 adsorption on LaB1−xB′xO3(001) (B, B′ = Mn, Fe, Co, and Ni, x = 0.0, 0.5). Then, charge analysis of the adsorption behavior is performed to understand the phenomenon in detail. Finally, the energetics of LiO2 adsorption is investigated to rapidly predict promising candidate perovskite catalysts. In order to tune the adsorption energy, which is closely related to the catalytic activity, we dope the B-site metal site with other 3d-transition metal species.24–26)
2. Computational Methods
All periodic density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP).27,28) The Perdew-Burke-Ernzerhof (PBE) functional, based on the generalized gradient approximation (GGA), was employed.29) In addition, the DFT+U method30) with Ueff = 4.0 eV (Mn), 4.0 eV (Fe), 3.3 eV (Co), and 6.4 eV (Ni)31) was applied to reduce the self-interaction error. All calculations included spin polarization and were performed with a cutoff of 400 eV. Geometries were optimized until the forces on each atom were below 0.03 eV Å−1. The optimized lattice constants were obtained using 2 × 2 × 2 supercells. An asymmetric slab structure was constructed by cleaving the bulk structure along the (001) plane. For the calculations of (2 × 2) surface unit cells with 4 atomic layers, an 8 × 8 × 1 Monkhorst-Pack32) grid k-point mesh was used. The slab models have two fixed bottom layers as the bulk positions and two fully relaxed top layers. A 15 Å vacuum layer was used to prevent interaction between the two periodic slabs. In all surface calculations, a dipole correction was applied in the perpendicular direction.33) We used BO2-terminated (001) surfaces, which often show high catalytic activity due to B-site metals.14,15) Fig. 1 shows the bare and doped perovskite surface structures used in this study.
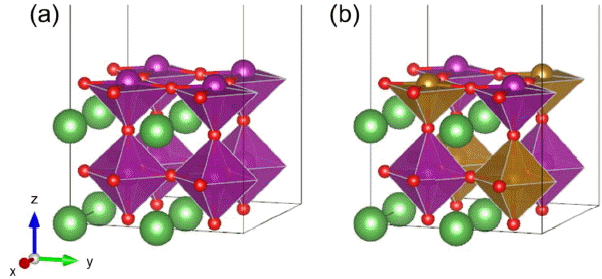
Unit cell of (a) LaBO3 and (b) LaB50B′50O3 perovskite surfaces. La (lanthanum) atoms are green spheres, B or B′ (transition metal) atoms are purple or brown spheres, and O (oxygen) atoms are red spheres.
The adsorption energy of LiO2 was calculated by
where
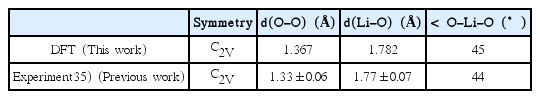
Comparison between Calculated and Measured LiO2 Geometric Information
Quantitative charge transfer analysis between adsorbates and surfaces allows us to deeply understand the adsorption phenomenon. Thus, an electron density analysis using the Bader charge model36,37) was carried out. A 12 × 12 × 1 Monkhorst-Pack32) grid k-point mesh was used for the Bader charge analysis.
3. Results and Discussion
3.1 LiO2 adsorption on LaBO3(001)
We first examined the adsorption phenomenon of LiO2 molecules on undoped perovskite (LaBO3) surfaces because these surfaces have relatively simple structures. Dathar et al. previously reported that the first e− transfer step of Li- ORR on a metal surface can be divided into associative and dissociative mechanisms depending on the activation barrier (Ea) for dissociating oxygen molecules.38) They suggested that a Li atom reacts with an oxygen molecule on metal surfaces with high Ea (LiO2 generation), whereas it reacts with dissociated oxygen atoms on surfaces with low Ea (OLiO generation). Unlike the case of metal surfaces, the initial adsorption mechanism on LaBO3 -type perovskite surfaces is still unclear. However, the O2 dissociation reaction often shows a high activation barrier (~ 1 eV) on A-site undoped LaBO3-type perovskites,39,40) which would have few oxygen vacancies due to the good balance of charge. This means that the first e− transfer step of Li-ORR on LaBO3 perovskite surfaces may be associative adsorption. Hence, we here examined the adsorption energies and site preferences of the LiO2 molecule, not the OLiO molecule.
Figure 2 shows the DFT-optimized LiO2 adsorbed LaMnO3 surface structure as a representative case. The O2 part in LiO2 is adsorbed on the top layer Mn atom. This is similar to previous reports in which the adsorption site of an O2 molecule on an ABO3 perovskite surface is the B-transition metal site.39–42) The Li atom in LiO2 is adsorbed on the 4-fold site surrounded by four lattice oxygen atoms. This adsorption site preference of LiO2 on the LaMnO3 surface was almost identical for the other LaBO3 perovskite surfaces examined in this study. However, the adsorption strength of LiO2 shows clear differences. The trend in the adsorption energies on LaBO3(001) (B = Mn, Fe, Co, and Ni) follows the order of atomic number of the B-transition metals in the periodic table (Table 2). This implies that the LiO2 adsorption energy is mainly affected by the properties of transition metals in the B-site.

(a) Side and (b) top views of LiO2-adsorbed LaMnO3 surface. La atoms are green spheres, Mn atoms are purple spheres, O atoms are red spheres, and Li atoms are blue-green spheres.

Adsorption Energies of LiO2 on LaBO3(001) (B = Mn, Fe, Co, and Ni)
3.2 LiO2 adsorption on LaB0.5B′0.5O3(001)
Our results for the undoped perovskites in Section 3.1 provide useful insight into how we determined the binding site of LiO2 on the B-site doped perovskite surfaces, LaB0.5B′0.5O3(001). As can be seen in Fig. 1(b), there are two possible adsorption sites according to the two types of transition metals exposed on the surface (B and B′). Based on the results of adsorption on the undoped LaBO3(001), we can reasonably assume that the O2 part of LiO2 is adsorbed on the top of B or B′ cations, while the Li atom in LiO2 binds only on the 4-fold site surrounded by four lattice oxygen atoms. We thus calculated the adsorption energies by considering those two possible adsorption sites for all LaB0.5 B′0.5O3(001) (Fig. 3 and Table 3).

LiO2 adsorption on (a), (b) Mn atoms and (c), (d) Co atoms in LaMn0.5Co0.5O3(001). (a), (c) indicate the side view while (c), (d) indicate the top view. La atoms are green spheres, Mn atoms are purple spheres, Co atoms are deep blue spheres, O atoms are red spheres, and Li atoms are blue-green spheres.
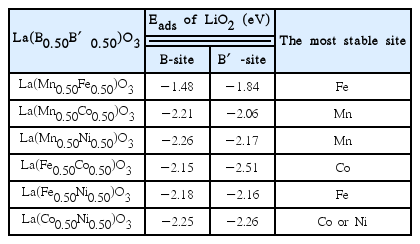
Adsorption Energies and Site Preferences of LiO2 on LaB0.5B′0.5O3(001) (B, B′ = Mn, Fe, Co, and Ni). The B- and B′-sites Mean the Adsorption Sites of O2 in LiO2
It is found that the adsorption energy and site preference of LiO2 on LaB0.5B′0.5O3 surfaces are not dependent on those characteristics of the undoped LaBO3 or LaB′O3 surfaces. For example, the adsorption strength of LiO2 (−2.05 eV) on the Co atom of LaCoO3(001) is larger than that (−1.75 eV) on the Mn atom of LaMnO3(001) (Because an Li atom is always absorbed at a site surrounded by four oxygen atoms, here we only mention the adsorption sites of the O2 part in LiO2). On the other hand, in LaMn0.5Co0.5O3(001), the adsorption strength of LiO2 (−2.21 eV) on the Mn atom is larger than that (−2.06 eV) on the Co atom. Furthermore, the adsorption strength on LaMn0.5Co0.5O3(001) becomes much stronger than that on LaMnO3(001) or LaCoO3(001). These unpredictable tendencies of the site preferences and adsorption energies of LiO2 imply that it is hard to minutely tune the adsorption energy (further, the activity) by doping or mixing of different metals in perovskite oxides. As a result, determining the fundamental origin of the characteristic adsorption phenomena must be definitively resolved and is a subject for further study.
3.3 Charge density difference analysis
Adsorption is often accompanied by charge density redistribution. Thus, to understand the adsorption phenomenon in detail, we analyzed the charge density difference using Bader charge analysis.36,37) Here, we demonstrate the results on LaMnO3(001) as a representative case. Fig. 4(a) indicates that charges of −0.20e, −0.06e, and −0.06e transfer from the LaMnO3 surface to the Ot, Ob, and Li in the LiO2 molecule, respectively (Here, Ot, and Ob indicate the atomic oxygens located at the top and bottom, respectively, of the adsorbed LiO2 molecule). Overall, −0.32e transfers from the surface to the adsorbate (LiO2). This tendency was also observed in the other perovskite surfaces we examined in this study. Ot has the most negative charge state and Ob and Li have relatively lower negative charge states after adsorption. Moreover, the total charge density differences of adsorbed LiO2 always have negative values; these values are related to the adsorption energies of LiO2. Fig. 4(b) shows the adsorption energies as a function of the changes in electron density on the ten bare or doped perovskite surfaces. Overall, the adsorption strength increases with increasing transfer of electrons from the perovskite surface to the adsorbed LiO2 molecule. These results indicate that the adsorption strength of the LiO2 molecule is mainly determined by electron charge transfer between the adsorbate and the perovskite oxide surface.
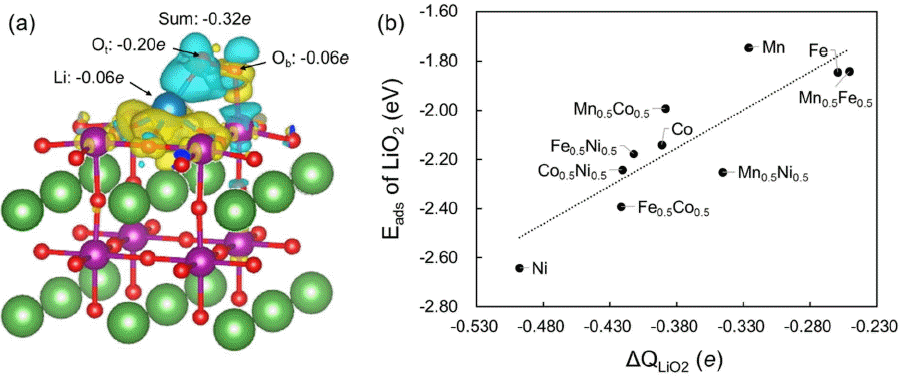
(a) Changes in the electron density upon LiO2 adsorption on LaMnO3(001). Yellow and cyan colors indicate decreased and increased electron densities, respectively. Ot and Ob indicate the atomic oxygens located at the top and bottom of the adsorbed LiO2 molecule, respectively. The isosurface contour is plotted with a charge density value of 0.002 e/Å3. (b) Adsorption energies as a function of the charge density differences on the LiO2 adsorbed LaB1−xB′xO3(001) (B, B′ = Mn, Fe, Co, and Ni, x = 0.0, 0.5). Each point indicates the pure or mixed B-site cations used in this system.
3.4 Significance of LiO2 adsorption analysis for Li-air battery cathodes
As mentioned earlier, LiO2 adsorption energy can be a useful descriptor for the activity because the weak adsorption strength of LiO2 is closely associated with the low overpotentials of ORR or OER.22,23) To estimate the activity of electrocatalysts for Li-air batteries, we assessed the adsorption energies of LiO2. Fig. 5 shows the DFT-calculated adsorption energies found in this study (reorganized from Tables 2 and 3). More red color means a weaker strength of LiO2 adsorption, while more blue color indicates stronger strength. Our results suggest that among the lanthanum based perovskites LaMnO3, LaMn0.5Fe0.5O3, or LaFeO3 would be good candidate materials as Li-air battery cathodes in terms of the reaction kinetics due to the weak adsorption strength of LiO2. Indeed, some of these materials such as LaSrMnO3 or LaFeO3 have already shown high electrochemical performance in Li-air batteries.16–18) This implies that our DFT results at the atomic scale provide a plausible explanation of the experimental results. To elucidate the detailed mechanism of ORR or OER in Li-air battery electrodes, a more thorough investigation for each elementary step will be required.

Adsorption energies of LiO2 on LaB1−xB′xO3 (B, B′ = Mn, Fe, Co, and Ni, x = 0.0, 0.5) perovskite surfaces.
In addition, unlike the case on metal surfaces,22,23,38,43) the adsorption phenomenon of Li-ORR or OER intermediates on perovskite surfaces have not been precisely determined at the atomic scale. Therefore, adsorption analysis of LiO2, a key intermediate of Li-ORR or OER, on a wide range of perovskite surfaces may provide useful insight into the adsorption study of the other intermediates.
4. Conclusions
In this study, we carried out structural, charge, and energetic investigations of LiO2 adsorption behaviors on LaB1−xB′xO3(001) (B, B′ = Mn, Fe, Co, and Ni, x = 0.0, 0.5) using DFT+U calculations. Unlike that of metal catalysts, the adsorption of lithium oxide intermediates on perovskite surfaces has hardly been examined. Our structural analysis showed that the adsorbed LiO2 has a similar adsorption site preference and structure in all BO2-terminated LaB1−xB′xO3(001). Charge analysis demonstrated that the charge transfer from the perovskite surface to the LiO2 molecule upon adsorption is a crucial factor for LiO2 adsorption. LiO2 adsorption energy was used as an activity descriptor to estimate the catalytic activities on LaB1−xB′xO3(001). Our results showed that LaMnO3, LaMn0.5Fe0.5O3, or LaFeO3 are promising candidate catalysts due to their weak LiO2 adsorption, even though the cathode performance is dependent on a number of other factors. The screening results we present here also offer useful insight for experimental research into a rational design of perovskite catalysts for Li-air batteries.
Acknowledgments
The authors acknowledge financial support from the Basic Science Research Program (2014R1A1A1005303) and the Global Frontier R&D Program of the Center for Multiscale Energy Systesm (NRF-2014M3A6A7074785), through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning; they would also like to acknowledge supercomputing resources, including technical support, from the Supercomputing Center/Korea Institute of Science and Technology Information (KSC-2015-C3-045).