Trends in Computational Materials Science Based on Density Functional Theory
Article information
Abstract
This review deals with computational treatments of subatomic levels of matter based on density functional theory (DFT), and tries to identify several current trends, which are largely consequences of the ever-increasing power of computers, which has substantially extended the performance of conventional DFT beyond its original scope. This review mainly focuses on the conceptual outline, rather than on lines of equations, highlighting several examples of calculations for each topic. It should be noted that these issues are hardly new to leading groups in the field, but certainly are for materials people in general. It should also be noted that the on-going efforts will continue and lead to a larger system size, a longer time scale, a higher accuracy, and a better efficiency of calculation for years to come.
1. Introduction
Watching raindrops on a lake, we may have a glimpse inside of nature as we consider the continuously decreasing positions of falling raindrops and constructive or destructive propagations of waves from the hitting points of raindrops on the water surface (Fig. 1). Indeed, our world seems to be made of two different worlds: the classical world of particles and the quantum world of waves. We know, however, that these worlds are just one single world built solely on waves. Sir Isaac Newton and people in his era and long after could not recognize the very tiny vibrations of particles. However, waves have been there for all matter since the beginning of the universe, the Big Bang. We now realize that what we observe with our limited vision in the visible-light range is just a simplified part of the whole wave world. Everything is waves after all.

Two worlds in one.
Thus a materials system can be described in two ways, classically and quantum-mechanically. In computational modeling and simulation, we simply try to bring a very small part of nature to a computer as a system, and apply the known rules of nature (Fig. 2) to solve certain problems at hand. The system may be an artificial one, and yet all the elements come from nature. So, the above definition of modeling and simulation is valid. And the two equations in Fig. 2, which became known when our country was in two periods of disturbance (the Jang scandal and the 6.10 movement, respectively) provide us what we are looking for.

This review deals with the latter, computational treatments of subatomic levels of matter, and tries to identify several new trends. This first principles (or ab initio) method is most accurate and relevant for materials studies, without any need for involvement of empirical parts like classical molecular dynamics (MD). In addition, although the rules of the quantum world are bizarre and cannot be derived, they have been justified up to the level of laws by their logical consistency and by the agreement of experiments over the years. They have never been disproved, nor has evidence been found to contradict them.
Whether we like it or not, our everyday life depends on hundreds of computers running unnoticeably in offices, banks, companies, stores, etc. They have changed everything from toys to spacecrafts and, most of all, the way we live. We often bring various realities on our computers instead of going out into the realities in person. Actions and counteractions take place on a real-time basis, instantly. Without a doubt, this tendency will continue if we look at the way our computing power is soaring upward (Fig. 3).

The subject of this review, current trends in computational material science using DFT (density functional theory), is of course directly related with this steady increase of computing power. More importantly, various combined approaches have been actively implemented in last ten years, such as DFT+hybrid functionals, DFT+U, DFT+ hybrid functionals+U, AIMD (ab initio MD)+hybrid functionals, etc. This review will treat these trends at an introductory level after a brief recapitulation on first principles methods.
2. Brief history of First Principles Methods
2.1 The beginning
Before we proceed to the main issues, it is worthwhile to look back briefly on how the entire field of computational materials science has developed. When classical mechanics, the “almighty” law of the universe for nearly three centuries, was found to be inadequate to explain phenomena on the subatomic scale, many scientists tried to put new findings together and formulate them into an equation. Schrödinger was the one who finally put it all together with the introduction of the wave function, Ψ.3)
This equation (see Fig. 2) can be praised one of the most ingenious ones ever written by a human being. At first glance, it is even innocent-looking, with two letters and two mathematical symbols in the form of a simple eigenvalue problem (Fig. 4). The energy Hamiltonian Ĥ (or energy operator) operates on the wave function Ψ, and yields EΨ. Like any other eigenvalue equation, Ψ remains Ψ, and energy E, which is what we are searching for, comes out as a constant. It has been stated that all information required to define a quantum state is contained in the wave function alone.

The whole process is like a magician (≡ operator Ĥ) taking a dove (≡ E) out of a supposedly empty hat (≡Ψ). We know that the dove was somewhere in there where we could not see it, and the magician simply moved her from the hat in a tricky way and showed the dove to us. There is no change to the hat due to this act. Similarly, all necessary information about E is contained in Ψ, and it shows up only after the operation of Ĥ. It should be noted that quantum acts are real things, whereas magic simply involves well-designed and manipulated illusions.
Solving the equation for more than two electrons, however, immediately leads to the n-electron problem, just like the N-atom problem in classical mechanics. When many bodies interact at the same time in a classical or quantum way, it is simply beyond our capability to solve exactly an equation for their interaction. The equation is useful in practice only for the simplest systems such as hydrogen-like systems. Scientists naturally engage in the usual routine, approximation. In fact, the development of the first principles method was through a series of approximations, especially for Ĥ and Ψ.
People often say that first principles methods require only a single piece of input data, the atomic number, and nothing else, for calculations. This is true, in principle, but the reality is not as simple as the statement sounds. The wave equation, in fact, is a partial differential equation that depends on the 3n coordinates of n electrons. We need to remember that we are talking about an amount of material of up to several hundred atoms, which could easily contain thousands of electrons. Then, devising and solving calculations for these n-electron systems are completely out of the question. This is the so-called “many-body problem.” Dealing with n electrons that interact with all other electrons at the same time is just too complex to solve, even numerically. Even if we knew how to do such a calculation, no computer that could handle such a massive task is available, and this will be true no matter how fast computers may become in the future.
The first move was to take the time variable out and assume that the heavier and slower nuclei are fixed on positions, resulting time-independent electronic Schrödinger equation. Hartree4) then approximated the n-electron wave function, Ψ, simply by multiplying electron’s wave functions each other, based on the result from the “electrons in a well” model. He further introduced the so-called one-electron model and a self-consistent procedure, both of which have been passed down in all the following methods as parts of the theoretical framework. Here, the one-electron model means that electrons are so near-sighted that they cannot see other electrons; they see only the mean-field created by other electrons. Of course, this method did not consider the quantum effect and yielded unsatisfactory answers, even when calculations were carried out self-consistently.
One of the keys to the calculation was the variational principle. Like the falling raindrops, our goal, a determination of matter’s ground-state energy, is always at the very bottom. We search for the global minimum in whatever way we can, until we reach a point of no further decrease in energy. With the wave equation, shown in Fig. 4, we vary the wave function until we hit the ground-state energy; this is called the variational principle. The principle allows us to start with a simple guess for the many-electron wave function.
Fock5) enhanced the Hartree method to a higher degree of perfection. This time, the key move was in the area of the wave function. In the Hartree–Fock (HF) method,5) the n-electron wave function is approximated as a linear combination of noninteracting one-electron wave functions in the form of a Slater determinant. And, the first quantum effect, the nonlocal exchange energy, came out from the determinant, satisfying Pauli’s exclusion principle at the same time. The HF method improved calculation results while maintaining the method’s parameter-free nature. However, practical application of the HF method is still limited to small systems with several tens of atoms, which are far from the normal regime of materials.
The energy operator Ĥ now consists of four terms for each electron; a kinetic term, a potential term between each electron and fixed nuclei, an electrostatic term between electrons, and a quantum exchange term of short-range repulsion between same-spin electrons. And yet, one more quantum term is missing and, for this reason, the energies calculated using the HF method are higher than those of data from accurately-performed experiments.
2.2 Density functional theory (DFT)
The breakthrough finally took place when Kohn and his two postdoctoral fellows, Hohenberg and Sham, published two dramatic papers. These changed the whole paradigm of wave function-based calculations, replacing the wave function with the idea of 3-dimensional electron density. The first paper by Hohenberg and Kohn, in 1964,6) proved that electron density at spatial points alone is sufficient to completely characterize the ground state of an n-electron system.
The second paper by Kohn and Sham, in 1965,7) described how the exact many-body energy can be calculated with a fictitious and non-interacting one-electron system whose energies are functionals of the electron density. This scheme introduced the exchange correlation (XC) energy, which contains all the interacting quantum effects, making the other energy terms free of such effect. The XC energy is formally exact and functional of the electron density. It is, however, unknown.
Again, an approximation is needed, and Kohn and Sham proposed the determination of XC energy by LDA (local density approximation),7) as schematically shown in Fig. 5. The many-body correlation effect comes principally from the short-range repulsion between electrons with antiparallel spins. It can be rather easily approximated from existing many-body calculations based on uniform electron gas (UEG) systems, completing now all terms including the correlation term for the energy operator Ĥ, as shown in Fig. 6. Schematically indicated in Fig. 6 as a background, electron density, the squared form of the wave function, decides everything in any n-electron quantum system, just as the wave function does. The successful debut of this LDA immediately initiated a rush of formulations of other XC functionals in the physics and chemistry communities.
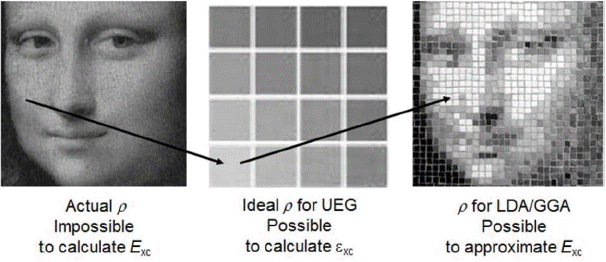

With the addition of gradient information on the electron density, GGA (generalized gradient approximation) functionals with PBE (Perdew, Burke and Ernzerhof) parameterization have been the default for solving the Kohn–Sham equations.8) And, the electron-density-based DFT (density functional theory) era started covering atoms, molecules, clusters, and materials. The calculated data are very close to data from accurately-performed experiments. Right after the announcement of GGA, people found that the best density functionals outperformed the best wave function methods and that DFT methods were computationally very inexpensive.
For materials, we introduced electrons into a vast terrain of varying potentials in a solid and try to identify the consequences. We eventually found a new formulation of the KS equations in terms of Fourier coefficients and witnessed the formation of band structures, a very important property of solids. To reach that end, however, two obvious issues still had to be resolved: the number of electrons becomes infinite, and so does the number of atoms in solids. Here, a series of smart algorithms and methodologies such as periodic boundary conditions (PBC), the supercell approach, pseudopotential construction for removal of core electrons from calculations, and k-point sampling with symmetry operations, etc. can greatly reduce the number of atoms, electrons, and calculation points.
At the end, we still maintain the physical relevance of real material and can treat bulk materials with a handful of k-points calculations. In addition, no matter how complicated a wave function is, we can decompose and reconstruct that wave function as one of superpositioned simple waves (plane waves, Gaussian waves, etc.) that can be easily handled by a computer, because the computational task now is to vary the weight factors until the calculated ground-state energy is minimized.
Although there is a problem in that either the system is still too small or the simulation time is too short for our purposes, the number of electrons that can now be calculated has increased up to several thousands with the advent of DFT, which changed from a 3n-dimensional equation to n separate 3-dimensional equations via the use of electron density. All properties, including electronic, magnetic, optical, and various response properties, can be obtained given that nuclei and electrons are considered in the method. These days, such methods have been so successful in both accuracy and efficiency that scientists are using them routinely in a wide range of disciplines, making computer experiments possible even for materials. One great thing about computational DFT is that absolute energy is not much of a concern. The important thing is the energy difference between two states or systems. This leads to error cancellation and makes the results more accurate.
3. Hybrid Exchange Correlation (XC) Functionals
In the DFT formulation, all terms are exact, with a sound basis in quantum mechanics, except for the exchange correlation (XC) energy. For this energy, there are troublesome and unknown quantum terms. Normally, this energy is less than approximately 10% of the total energy because the majority of interactions between nuclei and electrons are electromagnetic ones; however, XC energy is actively involved in determining important materials properties, such as bonding, spin-polarization, band gap formation, etc. As the name indicates, XC energy represents the lively activity of electrons among one another.
Thus, we have to approximate this energy as exactly as possible. The quality of a DFT run is critically determined by how closely the approximated XC energy reproduces the exact value. In fact, it is not a matter of great concern for us how XC functionals are generated. We are interested in which one is better than others, and in how much computational cost it requires. To determine these things, we have to understand the conceptual background of XC energy.
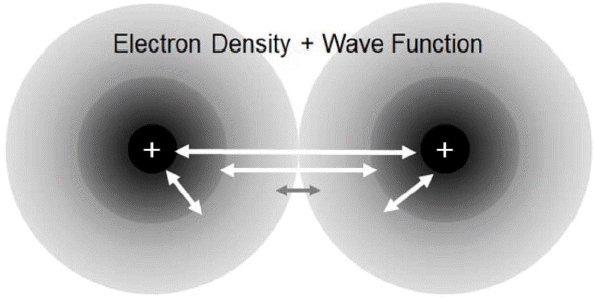
XC energy can be viewed easily with the introduction of an XC hole in an electronic system with two electrons, shown in the rough sketch in Fig. 7. It should be noted that the reference is the left electron. We expect that the presence of an electron will discourage the approach of another electron around it. As a result, there is an effective depletion of the electron density, namely, an XC hole, which has two components: exchange and correlation. These quantum interactions are interpreted as the electrons’ way of thoughtful consideration for others by “avoiding collisions and maintaining other’s privacy.” However, the Hartree energy covers the whole inter-electron distance, resulting in an overestimation of the repulsion. This should be corrected in terms of the XC energy, because the creation of XC holes around electrons leads to a net attractive energy.

Considering that actual systems are far from homogeneous electron gases, the LDA works fairly well in areas where the charge density varies relatively slowly, such as in covalent systems and simple metals. Its success was a surprise even to Kohn and Sham, who first proposed it. This is partly due to the cancellation of error in areas where LDA typically overestimates exchange energy while underestimating correlation energy. This implies that LDA satisfies the sum rule of the XC hole rather well, although each exchange or correlation hole does not. The spherically averaged value required for the energy calculation can be rather well represented as shown in Fig. 8.

Schematic of actual and LDA models for calculation of XC energy of H2 molecule.
However, whenever the situation deviates from the LDA or GGA models, it causes some errors and problems. An actual XC hole may have local variation such as degenerated energies and long-range tails (for example, in the case of a metal surface). Also, the XC potential generally does not accurately follow the asymptotic behavior (−1/r dependence) of the actual potential. The XC potential becomes too shallow near the nucleus and decays too quickly at long distances from the nucleus.
The development of new and more accurate XC functionals is a very active and ongoing field in computational materials science. This so-called “Beyond semi-local DFT/GGA” includes the following:
GGA with van der Waals interactions9)
GW approximation12,13) based on many-body perturbation of KS eigenvalue, which is especially popular in the solid-state community. It is now the method of choice for calculations of both the ground state and of quasiparticle band structures, as measured in direct and inverse photoemission. It is applicable in practice to molecular systems, open-shell systems, materials, and metals, providing very accurate band gaps, especially for weakly interacting systems such as semiconductors. However, it is still too computationally costly to be practical for general purposes, and is often used only as a reference calculation for materials.
Meta-GGAs
B3LYP, which is mostly used in computational chemistry.
Second-order Møller-Plesset theory (MP2), which is exclusively used in computational chemistry and belongs to the HF-based quantum chemistry approach.
Approach based on random-phase approximation (RPA), which deals with non-local correlations in a more systematic and non-empirical way.
Among these advanced developments, hybrid XC functionals and GW methods are the most relevant ones for materials. We limit our focus here only to hybrid XC functionals, especially the HSE06 functional,10) which has been very successful and is computationally affordable in materials calculations. HSE06 is designed to complement the conventional GGA/PBE functional by bringing in the HF exchange energy.
GGA functionals are approximated ones and suffer from self-interaction error, which leads to incorrect asymptotic potential curves. This error can be corrected by introducing range separation into the exchange component and by replacing the long-range portion of the approximate exchange by the exact HF counterpart. It should be noted that the HF exchange energy is wave-function based and is calculated non-locally. By incorporating non-locality into semi-local GGA functionals, we can eliminate most of the drawbacks of the GGA/PBE functional.
It should be noted that electrons move in an exchange potential screened by the electron holes of all the other electrons, and thus any process of screening is system-dependent. The screened hybrid functional HSE06 is a typical one; it consists of three terms: the HF exchange functional, the short and long range components of the GGA/PBE exchange functional, and the GGA/PBE correlation functional.
It seems that we are going backward to the HF model. We should remember, however, that the exchange energy as determined by the HF method is exact, contrary to the results of the DFT method; and, we have sufficient computing power to use this method, at least partially. The mixing portion depends on the screening degree; 25% mixing is just right for weakly-interacting semiconductors if vacuum is considered for a completely non-interacting case. Most DFT packages provide hybrid XC functionals, as schematically shown in Fig. 9, and normally require about ten new command lines in the input file including mixing parameters.
DFT model with hybrid XC functional for XC energy (short black arrows) between electrons engaged in quantum interactions.
Band gaps of most semiconductors and insulators calculated with this functional closely converge to the values determined by experiments and by GW calculations. This method also yields excellent results for vibrational properties, static and dynamic dielectric functions, and magnetic properties.14) In addition, the HSE06 results for Cu2ZnSnS4 as a potential photovoltaic material compare very favorably to experimental data for the lattice constant, optical spectrum, and band gap, all of which can be validated using G0W0 quasiparticle calculations.15)
The mixing parameter can be varied for different solids in relation to the degree of screening, the dielectric properties, or the electronic density.16) An example of successful application of a hybrid functional with a mixing parameter other than 0.25 is the case of ZnO.17) The theoretically derived defect energetics for ZnO always suffer from huge uncertainty between studies, no matter which calculation methods (including LDA, GGA, GGA+U, etc.) are applied. One of the main causes is the inaccurate description of the band structure of ZnO when using local or semi-local XC functionals. By using an HSE (a = 0.375) hybrid functional with finite-size corrections, defect energetics in ZnO consistent with the relevant experimental observations have been reported without resorting to empirical corrections for the valence and conduction-band positions.
It is thus true that hybrid functionals serve as a powerful tool and are practical enough methods for use in various computational fields. On the other hand, their applicability for cases of strongly-correlated d- and f- orbitals is still in doubt. Many transition metals and their compounds belong to this category. Recently, there have been various efforts to formulate a combined functional of hybrid and +U considerations together (The DFT+U approach is discussed in more detail in Section 5.).
Here, we are talking about the on-site occupation dependent XC energy and the effective potential of the hybrid functionals for localized states together. Ivády et al.18) connected both to the on-site correction term of the DFT+U method and indicated that the screening of the onsite electron repulsion is governed by the ratio of the exact exchange in the hybrid functionals. They claimed that the scheme provides a theoretical justification for the combination, and resolves issues caused by overscreening of localized states using tests for chromium impurity in wurtzite AlN and for vanadium impurity in 4H-SiC. There is no doubt that this level of functional will be added to the existing computational packages soon.
4. Click Computation
The title of this section has certainly not been favorably accepted by the physics or chemistry communities. For the materials sector, however, this could be something that many people have waited for, because they would like to have DFT computation as an accessible method of materials study. We often encounter runs that require a series of stages, such as calculations for band structure and DOS with hybrid functionals, phonon spectra and related thermodynamic properties, minimum energy path and barrier energy, interface properties between two phases, AIMD (ab initio MD), etc.
We also have to add the dispersion (van der Waals) effect, +U considerations, Bader analysis, and many other categories if the system requires it. In addition, post-processing based on the calculated data is an important step in computational work. All the above examples are quite time-consuming tasks. Especially in industry, engineers want to see results fast and understand the theoretical background later, only when needed. If something comes up, they can consult with theoretical and computational experts.
However, such results could be only clicks away if we employ a proper GUI (graphic user interface) package for these conventional and non-conventional DFT calculations. By using a GUI package loaded with all the essential and handy tools, we can complete jobs conveniently in much less time. Without a doubt, that will be a trend in later materials sector in the coming days and in the farther future. This kind of attitude toward computation used to be considered something that was not scientific. Not anymore.
With a GUI package, a series of tasks, stages, and subroutines can be automatically handled in a single job. All you need is to click, select buttons, and type simple parameters into boxes. It is like driving a car equipped with GPS (global positioning system), compared to normal driving without such a system. Fig. 10 shows typical GUI-based packages for computational materials science. These are undoubtedly the leading packages in the field, capturing all the features of DFT capability built into the same approaches: PBC, plane wave (PW) expansion of the KS orbital, and the pseudopotential (PP) scheme. It should be noted, however, that all of these packages have their own capabilities and merits. Unfortunately, all of them either require purchase or provide only limited trial offers.

Typical GUI-based packages for computational materials science.
Materials Studio19) deals with molecules, polymers, mesostructures, biomaterials, and materials. It is loaded with tools to handle quantum, atomistic, mesoscale, statistical, and analytical simulations. The package contains DFT methods (CASTEP20)), linear scaling DFT methods, QM/MM methods, semiempirical methods, MD, lattice dynamics, Monte Carlo-based methods, and force fields methods.
From the view of materials science, Materials Studio could be considered overloaded with too many tools, while MedeA21) is just about right, with all the essential tools for materials science, such as LAMMPS, Phonon, Surfaces, Interfaces, Nanotubes, Amorphous Materials, Transition State Search, electronics, etc. All inputs can be conveniently delivered via clicking or box filling, and results (band structure, phonon curves and corresponding thermodynamic data, electron density profile, potential profile, barrier energy profile for diffusion, Seebeck coefficient curves, etc.) are plotted or tabulated automatically.
For example, the package not only plots the phonon curves but also tabulates the thermodynamic properties automatically from the resulting data. Compared to performing the same tasks without it, the time saving is incomparable. It certainly is the best system for all users to achieve fast results.
Among the many useful tools in MedeA, the “Interfaces” tool, used to generate a two-phase system from two different phases, is one of the best we have seen. For example, exploring the interfacial property of WC-Co tool bits, two separate surface structures of WC and Co are needed, and an interfaced composite of WC-Co is also generated by combining the two prepared structures with minimal misfit.22)
5. Big Data
We are sensing and recording everything today. Our face, for example, is recorded more than hundred times every day and the image data are stored as many zeros and ones in memory devices. With the help of some intelligent program, the data can be screened, sorted, analyzed, and saved as a part of so-called Big Data. In any field, data sets are considered ‘big’ when they are large, complex, and difficult to process and analyze. These harvested sets of Big Data are accumulating in every sector of our society, every day.
The importance of this trend was manifested in 2012, when President Obama announced $200 million in new investment to support a Big Data Research and Development Initiative. This trend is again directly related to the increase of memory capacity and computing power. Computational data for materials are no exception. Every organization is involved in constructing modeling and simulating data in one form or another. Data for materials science tend to be particularly complex in terms of their type, phase, microstructures, etc.
In terms of sheer data volume, variety of data organization, velocity of data generation, and ease of data excess, the Materials Project23) led by UC Berkeley’s Prof. Gerbrand Ceder and Dr. Kristin Persson, has been the leader in the sector of computational materials science since they launched the project formally in 2011. Data and analysis tools are open to the public via web-based access requiring only one’s e-mail address. At present, the project presents a database on about 66,000 compounds, about 44,000 band structures, and much more. And of course, data on tens of new compounds are coming in every week.
For example, using the infrastructure of the Materials Project, scientists at the Department of Energy’s Lawrence Berkeley National Laboratory (Berkeley Lab)24) have provided the world’s largest set of data (on about 1,200 compounds) on the elastic properties of inorganic compounds.24) They claim that data on dozens more compounds are being added every week. This data set is especially valuable for materials scientists working on new structural materials, for which mechanical properties are the prime concern.
In addition, it has been claimed that a new thermoelectric material with low thermal conductivity was found in the screening process of the data. The material reportedly is significantly cheaper and more efficient at converting waste heat to electricity than any other material found to date. This clearly demonstrates the potential of Big Data for new materials development. As stated by Olle Heinonen of the Materials Science Division at the Argonne National Laboratory in Illinois, what matters most are our capabilities for processing data and ultimately getting something useful out of them.
Whether we like it or not, this Genome-style production of computational data is accelerating at full force like a runaway train. While one paper reports one single set of elastic data for a material, another project produces a whole spectrum of data, just like the automated production of aspirin pills. Even predictions of surface energies, point defects, and finite temperature properties using large data sets and advanced algorithms are underway. This is certainly a world in which the law of the jungle prevails, discouraging the participation of a minor country such as Korea. We have to accept this unstoppable trend as a fact of the era. However, instead of following the path of ‘Big Foot,’ we may first benchmark outside ideas, and then start based on the known data; we can then make every effort to find some niche points, either ones that others have missed, or one that we newly discover.
A smaller scale, but bold and significant, effort along these lines has been taking place at the School of Computational Science and Engineering at Georgia Tech, U. S. A., since 2014. With a grant awarded by the National Science Foundation of the U. S. A., the university is educating and training a new type of data scientist capable of creating advanced materials and bringing them to market at a fraction of the time, which at present generally takes many years. Based on materials informatics, this new graduate program at Georgia Tech will reportedly develop technologies to accelerate the design and manufacture of high-performance materials for applications ranging from fuel-efficient vehicles to emerging technologies such as 3D printing.
6. Calculations Beyond Conventional DFT
Accepting that the DFT/GGA approach covers a wide range of materials and their properties in practice, there is still a large class of systems for which the approximation of a KS single electron scheme is not valid. These calculations, once considered beyond the scope of DFT, have become routine tasks even for materials scientists these days. All DFT packages such as Quantum Espresso, VASP, MedeA-VASP, etc. have evolved continuously and are now armed with most of these new additions. Besides the methods that use hybrid XC functionals, discussed in Section two, there are the counting dispersion effect, GGA+U considerations, and AIMD, which are typical calculations that belong to this advanced category.
6.1. Addition of dispersion terms
The van der Waals (vdW) forces arise from dynamical polarization of the fluctuating charges of one particle with those in adjacent particles. DFT does not work for this case, becuase the method cannot provide a correct value of the −1/r6 dependence of the force with distance. Again, the reason is that the van der Waals force is a non-local correlation effect. The interaction is a seemingly weak one, but the dispersion effect is omnipresent and can add up to a substantial force in large assemblies. It thus can have a very strong influence on systems such as biomolecules, supramolecules, and nanomaterials.
The basic correction is as simple as that in which the additional energy is calculated pair-wise and added to the existing DFT energy. However, tens of different formulations are available in practice depending on the situation. For solids, the DFT-D2 method based on Grimme25) is the most popular; this method requires only two new lines (for example, LVDW = .TRUE. and VDW_SCALING = 0.75 for the VASP case). Normally, bulk solids do not require this correction, unless the system has a surface and molecules on it. Fortunately, the treatment of van der Waals interactions in DFT comes at essentially zero cost in terms of calculation time, because pair-wise calculations are rather straightforward.
6.2 +U
Transition metals are characterized by their partially filled d- and f-bands, and these localized electrons on the same atomic center create on-site Coulomb interaction, which leads to strong repulsion. This situation cannot be properly described with conventional DFT methods, which tend to delocalize electrons over the crystal and to make each electron feel an average of the Coulombic potential. The repulsion between these electrons is largely due to the contracted nature of the multiple charges, which leads to a greater correlation between their motions and to narrow widths for the bands associated with these electrons.
Because no method can capture electron exchange and correlation and eliminate self-interaction while remaining computationally efficient, we have to treat this electron localization problem separately and add to DFT, just as we added the dispersion effect. The DFT+U approach26,27) is the general practice for this, and uses an additional Hubbard-like term.
The strength of on-site interactions is usually described by parameters U (on site Coulomb) and J (on site exchange), or more frequently by a single effective U (= U − J).27) One study demonstrated the importance of using Hubbard parameters to model reaction energies involving transition metals even when no change in formal oxidation states was occurring. In actual runs, localized electrons are separated from other delocalized electrons (s- and p-electrons) and treated with the parameterized U as an HF-like potential. Because the correlation part is already counted in DFT, some double counting of the correlation part for localized electrons is subtracted from the Hamiltonian using another term.
This correction adds a penalty functional to the DFT total energy expression and forces the onsite occupancy into the direction of either fully occupied or fully unoccupied levels. Normally, effective U parameters are provided by referring to the first principles (or ab initio) calculations, employing a hybrid functional or unrestricted HF method, or are fitted to the experimental binary oxide formation energies.28) It should be noted that the on-site interaction energies must be constant for any given material.
Selecting the right U parameter for a given system can require a survey of the literature survey and preliminary trials, because such a process is partly empirical in nature and must sometimes be adjusted to obtain the best experimental data. This type of GGA+U calculation can be easily implemented in the VASP package, for example, with only seven additional lines in the INCAR file. However, the calculation time is about 1.5 times that of the conventional DFT calculation. It should be noted that, by pushing d-bands away from each other due to a repulsive penalty, the GGA+U method yields higher energies and wider band gaps.
6.3 ab initio MD (AIMD)
Only ten years ago, materials scientists mostly hesitated to run AIMD because of the high computational cost; they turned to classical MD or simply gave up. The situation has changed now, and many use data to study the time evolution at temperatures higher than the normal 0 K, even though the cost is still high. Once again, the enhanced power of computers is behind the scenes making these kinds of jobs practically possible. DFT-based molecular dynamics (MD) methods, such as Car–Parrinello MD (CPMD)29) and AIMD,30) have enjoyed a rapidly increasing popularity in the past decade, and are now ‘routinely’ applied in many areas of materials science.
DFT-based MD simulations allow a more realistic description of material systems and their processes, with a full description of the dynamical ensembles at elevated temperatures; these simulations are thus mimicking actual experimental conditions ever more closely. One such example revealed localized electrons (polarons) on the surface of hematite via a calculation using the hybrid DFT of AIMD, while normal AIMD predicted a delocalized electron.31) For transition elements with tightly bound electrons in oxide materials, a combined approach was found to be needed and a highly scalable algorithm for exact exchange was developed and incorporated into this AIMD run. One conclusion is that the lower levels of XC potentials presently used in AIMD simulation cannot be used to reliably predict the properties of many materials. Expanding the length and time scales that are accessible with AIMD will remain a challenge and a topical area of research in years to come.
Property calculations are not used anymore for bulk properties only. These days, runs largely aim at multi-phase systems, interfaces, devices, etc., which are very relevant and important for materials study. The following are typical examples of application-oriented runs on materials based on DFT:
7. Remarks
In computational materials science, we aim to understand the various properties and phenomena of materials and to achieve the design and fabrication of better materials for society. This goal is realized by modeling materials with computers that are programmed with theories and algorithms based on physics, mathematics, chemistry, materials science, and computer science. In many cases, a computational approach may be the only way to handle materials under extreme and hostile conditions that can never be reached in a laboratory, such as under high pressures, at high temperatures, and in the presence of toxic substances or nuclear radiation. For example, the materials in nuclear fusion environments are of great concern these days. The various damages occurring in fusion materials due to neutron irradiation can be simulated without worrying about expensive equipment or the danger of radiation.
DFT, the leading first principles method, is replacing a large portion of trial-and-error experiments with computationally prearranged ones. As a result, average R&D workload has dropped by about 30%, and the steady upgrade in computing power is telling us that this drive will continue. Thomas Watson, the former chairman of IBM, surely would like to take back his original statement and restate it as “I think there is a world market for everyone except for maybe five people.” It should also be noted that Moore’s Law is still valid.
Presently, the materials sector accepts computational science as a handy and useful tool for experimental work like XRD, SEM, TEM, STM, etc. Modern DFT codes can calculate a vast range of structural, chemical, optical, spectroscopic, elastic, vibrational, and thermodynamic properties, and it is nowadays common practice to include computational results in experimental studies on materials. It is not necessary to understand all the theories, algorithms, and equations in detail, nor the codes or programs down to the bits and bytes. Such knowledge is reserved for other professionals such as physicists and chemists. If we embrace this fact and do some sorting and trimming of the subject, we may be able to venture into the field without too much difficulty. It should be noted that, presently, about 30% of people in the field of computational materials are from areas other than the computational fields. Computers are here to serve us, and their limits might be our imagination and our imagination only.
Acknowledgements
The author would like to acknowledge the support by Materials Design (www.materialsdesign.com/medea) and Kyungwon Enc. (www.kwenc.kr) for granting the use of MedeA-VASP program.