1. Introduction
As a major bone component, calcium phosphates (CaP) for the source of hydroxyapatite (HAp, Ca5(PO4) 3(OH)) have been widely used in the production of artificial materials that can substitute for autogeneous bone.1-2) For the purpose of improving the material’s toughness, we have attempted to develop CPC (Calcium Phosphate Compound) bone cement. Since the first CPC, consisting of an equimolar mixture of tetracalcium phosphate3-4) (TTCP) and dicalcium phosphate anhydrous (DCPD), was developed in 1986, many kinds of CPC powders have been developed, mainly by using TCP and/or TTCP.
β-TCP is the low-temperature phase in the CaO-P2O5 phase diagram,3-4) and is known to be resorbable in vivo with new bone growth, replacing the implanted β-TCP.5-7) Tricalcium phosphate is one of the most important biomaterials based on phosphates, and is currently recognized as an artificial bone material that significantly simulates the mineralogical structure of bone.8-10) Theoretically, resorbable TCP is an ideal implant material. After implantation, TCP will degrade with time and can be replaced with natural tissues. This process leads to the regeneration of tissues instead of their replacement and, as such, solves the problem of interfacial stability.11-12) Often, tribasic calcium phosphate is mistaken for β-tricalcium phosphate. According to Metsger et al.,12) tribasic calcium phosphate is a nonstoichiometric compound often bearing the formula of hydroxyapatite [Ca10(PO4)6(OH)2]. TCP is a resorbable temporary bone space filler material.
We have reported the preparation of a nonstoichimetric Calcium Phosphate Compound [CPC].13-15) In previous research, the nano-crystalline calcium phosphate (CaP) paste was similar to the β-TCP phase; our material, bio-TCP (B-TCP)5,6) was prepared at 37°C in an aqueous solution of active Ca(OH)2 and H3PO4. In order to increase the toughness of the artificial bone using β-TCP,16-20) we have focused on the crystal growth of the B-TCP crystallites and the mixing effect of the crystallites with various dimensions. The crystal growing of B-TCP crystallites and the mechanical strength of the TCP mixture blocks are reported.
2. Experimental Procedure
2.1. Precipitation of β-tricalcium phosphate phase using Ca(OH)2 and H3PO4 in water
Figure 1 shows the preparation process and the characterization of β-TCP powders and artificial bone blocks. From the phase diagram3-4) of [Ca2+] and pH, it can be seen that the β-TCP phase is formed at pH 5.0. The calcium phosphate (CaP) precipitates were prepared in a CO2-free air atmosphere by a conventional wet method13-15,17) using Ca(OH)2 and H3PO4. The starting materials were CaCO3 (Alkaline analysis grade, Aldrich, USA) and H3PO4 (AP grade, Aldrich, USA). As precursors for Ca2+ and H3-xPO4 ions, we used highly active Ca(OH)2 and H3PO4 in DI water. The highly active Ca(OH)2 was obtained through the hydration of CaO, in which CaO was prepared by the calcination of CaCO3 at 1150°C for 4 h. The CaO, obtained at 200°C, was hydrated using 3 mol% of DI H2O to make Ca(OH)2 powders; resulting mixture was stirred in 1000 cc of DI water to make the ionized aqueous solution of Ca2+. The obtained ionized Ca2+ solution was used as a base for the titration process. The calcium phosphate slurry was prepared using the simultaneous titration method with peristaltic pumps (Masterflex, Cole-Parmer, USA), a water bath (Boekel, USA), and a pH controller (Bukert 8280H, Germany). The temperature of the main reactor was digitally controlled to within 0.1°C. During the precipitation, the reaction solution in the vessel was maintained at 37°C and at pH5.0±0.1 under stirring. The CaP slurry pastes were denoted as B-TCP37.
2.2. Crystal growth of bio-tricalcium phosphate slurry paste and fabrication of β-TCP blocks
For the crystal growth of nano-precipitates in the CaP slurry pastes, B-TCP37 slurry solution in a beaker sealed with vinyl film and kept in the drier at 90°C according to the time schedule from 12 h to 72 h; the obtained CaP solution was filtered and the CaP cake was dried at 90°C. The obtained dry powders were denoted as B-TCP90-12, B-TCP95-24, B-TCP90-36, B-TCP90-48, B-TCP90-60, and B-TCP90-72 according to the time schedule of 12 h, 24 h, 36 h, 48 h, 60 h, and 70 h, respectively. Especially, the precipitation of the B-TCP95-12 sample was performed at 37°C in a beaker under stirring; precipitation was sustained at 95°C overnight without stirring until the next day.
After the precipitation, some of the CaP slurry pastes were filtered using an aspirator and dried at 90°C overnight in order to obtain dry powders, which were then characterized using XRD and FE-SEM.
In order to investigate the solid state interaction5,6,17) among the CaP powders in a mixture block of B-TCP37 and B-TCP90, B-TCP37 nano-crystalline powders were uniformly coated on the surface of the B-TCP90 macro-crystalline crystals using a vibrator (FRITSCH, analysette 3 SPARTAN) and the mixture was calcined at 1150°C for 4 h. The goal of this process was to obtain a uniform mixture block of nano-crystalline B-TCP powders with macro-crystalline B-TCP90 powders. The mixture powders were put into the mold and vibrated using a FRITCH vibrator for 10 seconds; then, the mold was reversed and vibrated again for ten seconds. This process was performed three times. Finally, the mold was put on the press and hydrostatically pressed at 12 tons for 10 seconds. The pressed sample blocks were fired at 1150°C for 4 h. The mixing amounts of B-TCP37 powder to 2 g of B-TCP90-72 powders were in a range from 0.5 g to 5 g.
2.3. Characterization
The crystal phase was confirmed using X-Ray Diffraction (Bruker, M18XCE) of the crushed powders. XRD patterns were obtained for the commercial β-TCP sintered nano-powders at sizes of 150 nm [Sigma-Aldrich, 16483]. The preparation process of β-TCP nano-powders sensitively affects the formation of crystal phases. In this research, commercially well-known β-TCP powders were used as a reference for the comparison of crystal phases.
The morphology of the CaP powders and the CPC samples was observed using FE-SEM [Hitachi, S-4800]. The compressive strength values of the formed and calcined TCP blocks were measured using a universal testing machine (UTM, DaeSan Eng, Korea). The sample size of the TCP block was 2.7 cm Φ × 8 mm t after firing at 1150°C for 6 h. For the TCP block sample, several tens of μm β-TCP90 powders were mixed with the proper amount of nano-CaP powders, which had dimensions under 100 nm. The mixing ratio of the B-TCP37 powders to the B-TCP90-72 powders was from 10 wt% to 50 wt%. At levels above 50 wt% of B-TCP37, the compressive strength approached a saturated value. The B-TCP90 powders were mixed with B-TCP37 powders using a vibrator and compressed into a mold using a hydraulic press to make the TCP block. The formed mixture block was calcined at 1150°C for 4 h.
3. Results and Discussion
3.1. Preparation of β-TCP granules and block
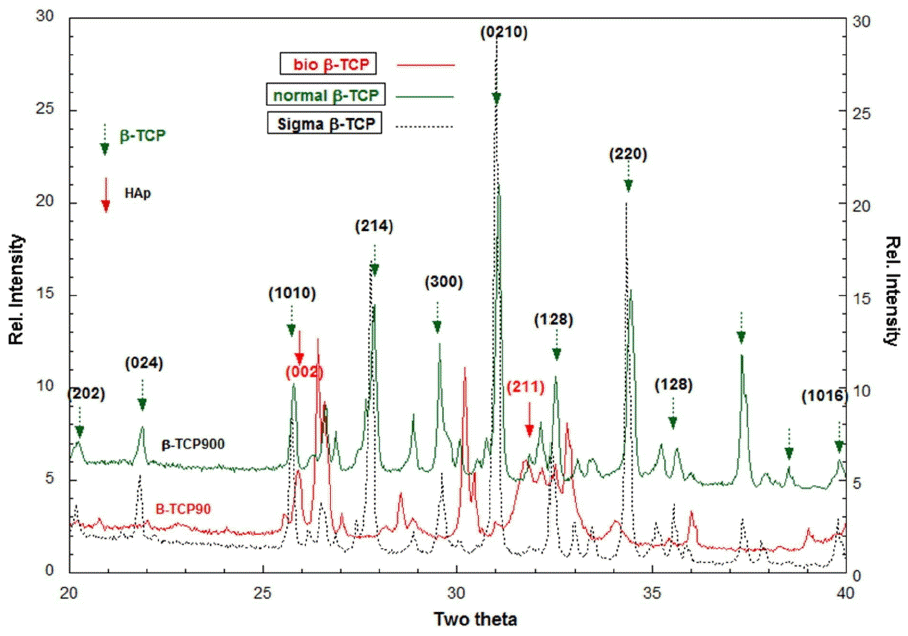
In Fig. 2, the main phases of B-TCP37, which were prepared at pH 5.0 and 37°C, are identified as apatitic TCP (Ca9(HPO4)(PO4)5OH); these results were obtained through XRD analysis. The obtained slurry solution, B-TCP37, was sustained at 90°C according to the time schedule for the crystal growth. The XRD spectra for Bio-TCP90 [B-TCP90] is close to that of apatitic β-TCP,18) and β-TCP900 shows typical XRD spectra, close to those of commercial β-TCP nano-powders [Sigma-Aldrich]. That is, the apatitic TCP was changed to β-TCP phase through the calcination at 900°C because the mole ratio of Ca2+ to PO43− was 3/2 in the wet precipitation. The B-TCP900 showed normal β-TCP crystallites, in which B-TCP37 was dried and calcined at 900°C to make B-TCP900 powders.
Figure 3 shows typical crystal growth for the samples of B-TCP95-12, B-TCP90-36, B-TCP90-48, B-TCP90-60, and B-TCP90-72. The crystallites were rectangular in shape, and so the crystal dimensions were identified as L, W, and T, respectively. In Fig. 3(a), the dimensions of B-TCP-37 were estimated at L = 200 nm, W = 30 nm, and T = 20 nm; the crystal size greatly increased to several or several tens of μm with the increase of the crystal growth time. The FESEM microstructures in the figures from Fig. 3(b) to Fig. 3(f) show normal crystal growth of TCP hexagonal crystallites with reaction time and reaction temperature.19-25) It should be noted that the B-TCP95-12 sample slurry was prepared at 37°C and kept for 12 h at 95°C. B-TCP95-12, as can be seen in Fig. 3(b), shows bigger crystal growth induced by the higher reaction temperature, compared with the crystal growth of B-TCP90-36.
For the increase of mechanical strength of β-TCP bone block, nano-crystalline B-TCP37 powders were blended with macro-B-TCP crystallites, which were prepared by wet process. Fig. 4 shows the grain growth in the β-TCP mixture block, which was prepared through mixing and firing of B-TCP37 powders and B-TCP90 powders. Through vibrating mixing, the nano-crystalline B-TCP37 powders, as can be seen in Fig. 3(a), were well attached to the surface of the uniformly arranged B-TCP90 macro-crystallites, as can be seen in Fig. 3. In the FE-SEM microstructures shown in Fig. 4 we can observe nano-size grains of 0.1 μm order and macro-size grains of μm order. In Fig. 4(a), there are sub-micrometer crystallites in the blue round circle; we can observe large grains of ~5 μm. Laminated grain growth of the crystallites can be observed in the red round circle in Fig. 3(a) and 3(b). As mentioned in the experimental section, the macro-B-TCP90 crystallites shown in Fig. 3 were uniformly arranged with the nano-B-TCP37 crystallites in a mold using a FRITCH vibrator; the mixture was pressed using a uniaxial press. When we mix the rectangular macro-crystallites with the nano-crystallites powders, the resulting structure looks like a wall construction using red blocks and cement paste. It is supposed that the mixture of parallel arranged crystallites induced the laminated grain growth during the sintering at 1150°C for 4 h. Furthermore we may control the microstructural morphology of the β-TCP block via the lamination reaction of grains during the sintering schedule; this will increase the mechanical strength of the β-TCP bone block.
Figure 4(g) shows the microstructure of the sintered mixture block of B-TCP90-48 with B-TCP37. For the porous bone block, the B-TCP mixture was prepared through the mixing of 50 wt% B-TCP90, 50 wt% B-TCP90-48, and polystyrene beads for porosity control. The B-TCP mixture was shaped and then the shaped block sample was fired at 1150°C for 4 h. The pore size of the β-TCP-block was estimated at ~280 μm. In Fig. 4(c), submicron-grains (0.1-0.3 μm) can be seen to have formed via solid-state interaction among nanometer crystallites such as B-TCP37; bigger grain growth (5 - 7 μm) was found to have occurred via the condensation reaction among macro-crystallites [B-TCP90] and nano-crystallites [B-TCP37]. Fig. 4(e) shows crystallite granules in the circled regions, in which the several tens of nm B-TCP37 powders were grown to nearly μm size through coagulation and grain growth. Two yellow round circles also show the condensation region formed by the solid state interaction among nano-TCP granules. Fig. 4(f) shows the β-TCP grain growth, induced by the nano-B-TCP37 crystallites. The blue circle region in Fig. 4(g) indicates a β-TCP matrix grain and, in the yellow circle region in Fig. 4(h), one micrometer β-TCP crystallites are interconnected with several tens of micrometer β-TCP matrix grains. In Fig. 4(h), the yellow circle region corresponds to Fig. 3(d).
3.2. Mechanical property of β-TCP block
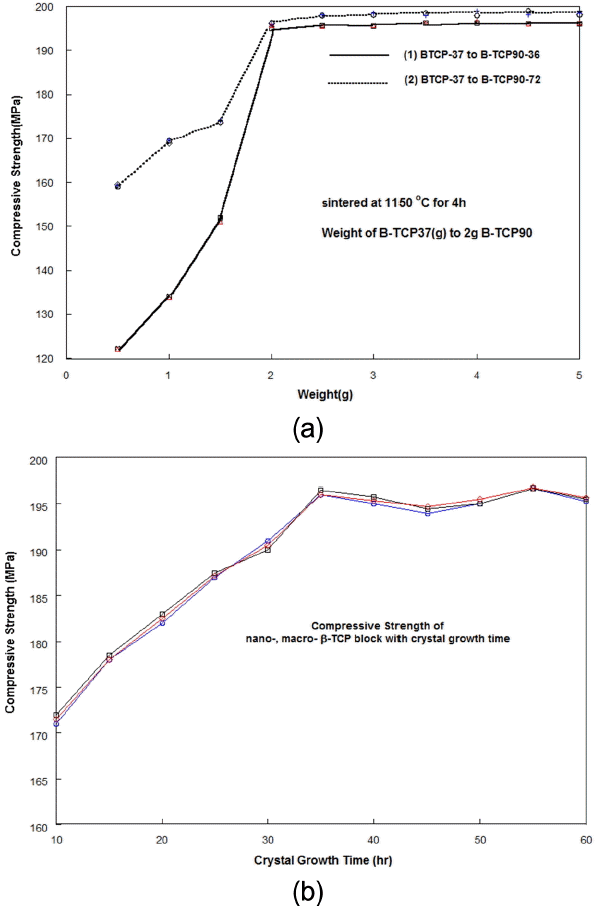
There are two key factors in the density of artificial bone and in the improvement of the deflection. The blending technology of nano-B-TCP37 and macro-B-TCP90 can be used to increase the deflection behavior of an artificial bone block. Fig. 5(a) shows the compressive strength for the mixture blocks, in which the first group samples were prepared using B-TCP37 and B-TCP90-36, and the second group samples were prepared using B-TCP37 and B-TCP90-72. The powder mixing ratio of the block samples varied with the weight of B-TCP37 to 2 grams of B-TCP-90 powders. The shaped mixture block was sintered at 1150°C for 4 h. The compressive strength of sample group (1) is much bigger than that of sample group (2), below the value of 2 wt% of the B-TCP37 amount. Above a 2 wt% amount of B-TCP37, the compressive strength for all test specimens approached the saturated value. The compressive strength values of the group one blocks using B-TCP90-36 were lower than those of the group two blocks using B-TCP90-72. From these results, it is supposed that if we increase the crystal growth time of B-TCP90, the compressive strength value may be much higher. We will discuss these results in the next report. Fig. 5(b) shows the compressive strength with the variation of crystal growing time in the B-TCP90 samples. It should be noted that the compressive strength values approached the saturation value above 36 h of crystal growth time of B-TCP90 in spite of the variation.
According to one previous report13-14) on β-TCP based bone blocks, the mechanical strength was much less than that of real bone; one of the reasons was the difficulty of density control. In this report, we have tried to prepare uniform microstructures using macro- and micro-crystallites having rectangular shapes. Several or several tens of μm crystallites in B-TCP-90-36, B-TCP90-48, B-TCP90-60, and B-TCP90-72 were coated with several tens of nm crystallites of B-TCP37. We wanted the hexagonal crystallites in the macro-crystalline B-TCP90 to be well arranged and uniformly coated with B-TCP37 nano-crystallites.
The artificial block of macro- and nano-crystalline TCP composite showed a better mechanical property, which was confirmed by observation of the microstructural integration among the crystal surfaces. In this study, to investigate the mechanical property of the mixture blocks of macro- and nano-B-TCP crystallites, we have tried to control the dimensions of the wet crystallites, prepared at pH5.0 and mixed with nano-crystallites. The tensile strength increased gradually with the mixing ratio and the dimensions of the macro-crystallites, which were prepared at 90°C. Using UTM, the tensile strength of the β-TCP mixture block was evaluated at ~ 12.6 MPa; this value increased with the increase of the amount of B-TCP37. The details of the tensile strength with toughness will be reported in the next issue.
4. Conclusions
When the nano-B-TCP37 powders were mixed with macro-B-TCP90 powders, the fired TCP blocks showed higher mechanical strength. In the mixture blocks of nano-B-TCP37 and macro-B-TCP90-72, the wt% mixing ratio of 50 to 50 showed the highest compressive strength of 196 MPa. The crystal growing temperature and the time greatly affected the compressive strength of nano the - and macro-β-TCP mixture block.