1. Introduction
In dentistry, a number of synthetic biomaterials are used for the replacement of destroyed or lost structures and for the restoration of disturbed functions of the orofacial organ.1) Dental cements are such a class of biomaterials that are widely used in restorative dentistry.2,3) Inorganic phosphates, 4) silicate cements,5,6) glass ionomers and their composites, 7,8) and resins9) are the major biomaterials that are used as dental cements. Among the various silicate cements, hydraulic calcium (alumino)silicate cement, more commonly known as mineral trioxide aggregate (MTA), is an alternative biomaterial for dentine replacement, pulp capping, pulpotomy, creation of apical barriers in teeth with open apices, and repair of root perforation and resorptive defects as well as orthograde or retrograde root canal fillings.10-12) MTA is a clinker-derived Portland cement composed of different phases, including tricalcium silicate, dicalcium silicate, tricalcium aluminate, tetracalcium aluminoferrite, and calcium sulfate. 13-15) However, its higher cost, handling difficulty, and longer setting time are the major impediments to its widespread use.12)
Calcium silicate-based Portland cement (PC)-type materials have gained popularity in recent years owing to their resemblance to MTA, and various clinical applications such as root perforations, apexification, resorptions, retrograde fillings, pulp capping procedures, and dentine replacement have been recommended for them.16-18) However, the presence of toxic heavy metals in PC is one of the important aspects in the development of PC-based low-cost dental materials.19-21) To formulate a low-cost material based on MTA or Portland cement with a shorter processing time and minimal cytotoxic implications, several compositional manipulations have been made to the existing commercial dental cements.22-27) In an attempt to develop new endodontic cement from pure raw materials, which has the advantages of both MTA and PC at a lower cost, we had prepared an experimental PC and investigated its physical properties and biocompatibility.28,29)
The water dynamics and kinetics of the setting process of calcium silicate-based dental cements are complex processes and highly influenced by the composition, especially that of calcium trisilicate.6,12,30) In the present work, to determine the role of individual constituents—tricalcium silicate (C3S), dicalcium silicate (C2S), and tricalcium aluminate (C3A)—in the setting process, we first investigated the hydration process of these constituents. Then, to develop new compositions of PC-based dental cement, we prepared four different compositions of experimental PC (EPC) by mixing pre-formed C3S, C2S, and C3A, which are the main constituents of PC,12) in different ratios and investigated their physical properties, setting process, and biocompatibility. The results were compared with those of a commercial dental cement, ProRoot MTA, and an experimental cement (EPC) prepared directly from the pure raw materials.29)
The cement paste formed during the setting process is composed of a continuously altering interconnected pore network filled with mobile ion-containing water, which makes it electrically conductive, and the changes in its magnitude with time during the setting process was monitored in situ.31,32) Electrochemical impedance spectroscopy (EIS) is one of the major techniques for the in situ monitoring of hydration process of cement-based materials.33-35) In the present study, we performed frequency-dependent impedance spectroscopy for the in situ monitoring of hydration process of various compositions. Furthermore, the cytotoxicity of various compositions was investigated by performing XTT assays, and the results were compared with those of the commercial dental cement, ProRoot MTA.
2. Experimental Procedure
2.1. Synthesis of C2S, C3S, and C3A
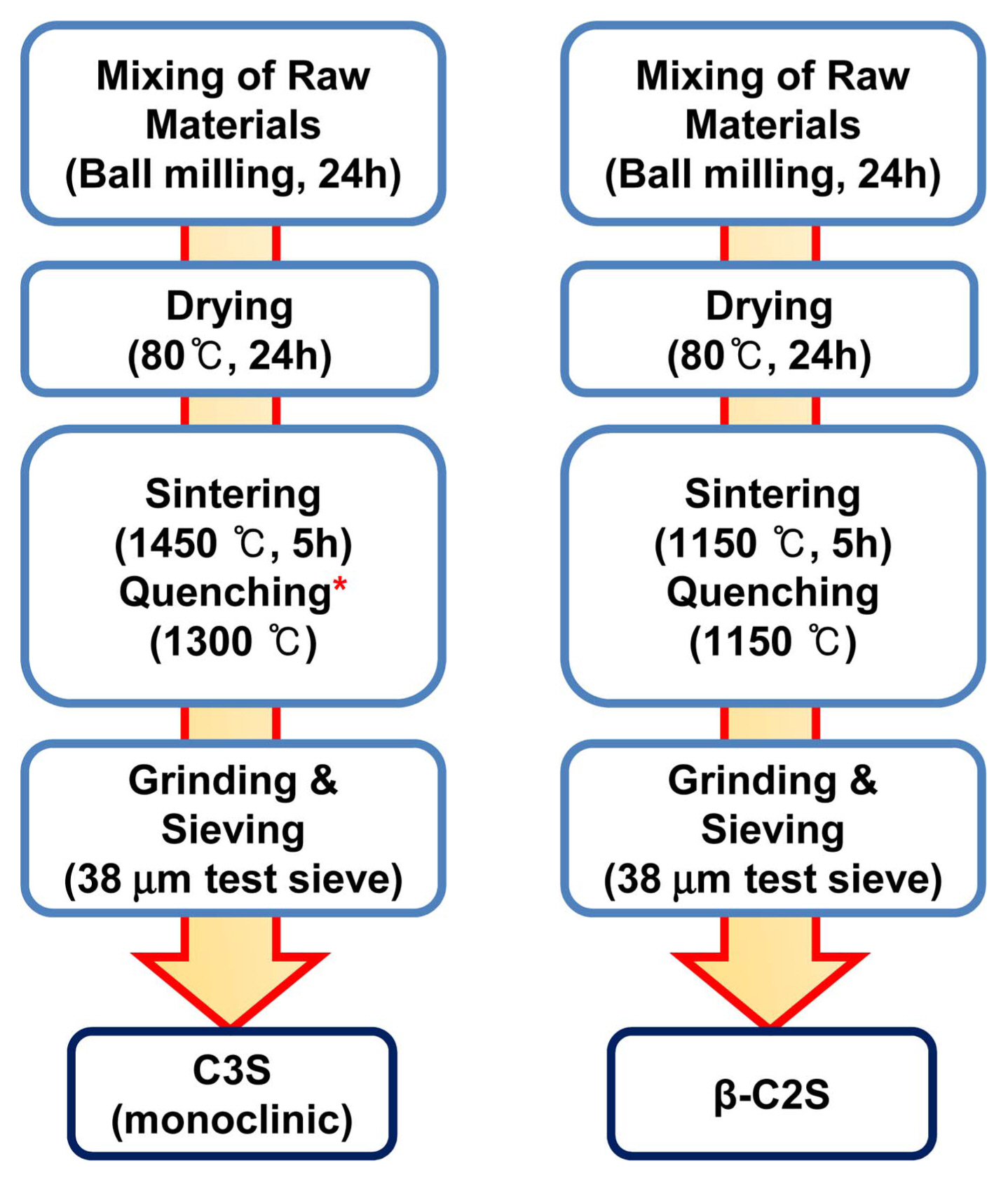
C2S and C3S were first synthesized by solid-state reactions between pure raw materials; the purity and mixing ratio of the various raw materials used in the study are listed in Table 1. Fig. 1 shows the flow chart of the various steps involved in the synthesis of C2S and C3S. Briefly, for each composition, the raw materials were mixed in an appropriate ratio, as mentioned in Table 1, and ball-milled in isopropyl alcohol (IPA) for 24 h. The ball-milled mixture was dried in a hot-air oven at 80°C for 24 h. A semi-dried slurry was prepared by moistening the dried powder with 15 wt% IPA. Spherical balls of 1.5 cm diameter were formed from the semi-dried slurry and dried in a hot air oven at 80°C for 1 day. The dried spheres were sintered in air at different temperatures and then quenched in liquid nitrogen. The heating rate during sintering was fixed as 3°C/min. The clinker obtained after quenching was fully ground and sieved using 38 μm mesh test sieves to obtain the desired C3S or C2S powder.
C3A was prepared by the Pechini method36) using nitrate salts, Ca(NO3)2·4H2O and Al(NO3)3·9H2O, as precursors. The aqueous solution of the precursors was prepared by dissolving them in ~ 50 ml of deionized water. Then, citric acid was added and the mixture was stirred to obtain a clear solution. Ethylene glycol was added to this solution, and the solution was continuously stirred at 80°C, at which polyesterification reaction occurred along with the evaporation of excess water, which resulted in the formation of a viscous gel. The gel was dried at 150°C in a hot-air oven for 24 h. The dried gel was heated at 400°C for 3 h and then ground to a powder form. A semi-dried slurry was prepared by moistening the dried powder with 15 wt% IPA and spherical balls of ~ 1.5 cm diameter were formed. The dried spheres were sintered in air at different temperatures for 4 h and then quenched in liquid nitrogen. The clinker obtained after quenching was fully ground and sieved using 38 μm mesh test sieves to obtain the desired C3A powder.
2.2. Synthesis of experimental Portland cements (EPC) from C2S, C3S, and C3A
EPCs with four different compositions were prepared by mixing pre-synthesized C2S, C3S, and C3A with gypsum in different weight ratios, as given in Table 2.
2.3. Structural characterization
The phase compositions of C2S, C3S, C3A, and various EPC compositions in the powder form as well as during the hydration process were analyzed using a high-resolution X-ray diffractometer (D/Max Ultima III, Rigaku, Japan) equipped with a Cu-Kα radiation source (1.5406 Å) at a scan rate of 1°/min in the scanning angle (2θ) range of 10°-80°. Phase identification was carried out by matching the experimental XRD data with the Inorganic Crystal Structure Database (ICSD) of Korea Institute of Science and Technology Information. The microstructures were analyzed using a field-emission scanning electron microscope (FE-SEM, S-4700, Hitachi).
2.4. Cytotoxicity test
The cytotoxicity of the EPCs and ProRoot MTA was determined by performing an XTT assay. Human dental pulp cells (HPC) and human immortalized dental pulp cells (HIPC) were seeded at 1 × 104 cells per well on 96-well cultures for 24 h and treated with MTA (ProRoot MTA; Dentsply, Tulsa, OK) and the EPCs’ extract solutions for 24 h. After 24 h, the cell viability was analyzed using an EZ-Cytox enhanced cell viability assay kit (Daeil Lab Service, Seoul, Korea). Briefly, 10 μl of EZ-Cytox reagent was added to each well of the 96-well plate and incubated at 37°C for 4 h. The absorbance was measured at 420 nm using a spectrophotometer (VERSAmax multiplate reader; Molecular Devices, Sunnyvale, CA).
2.5. Electrochemical measurements
The electrochemical analysis of the hydration processes of C2S, C3S, C3A, and various EPCs was performed by EIS. The specimens for EIS were separately prepared in a specimen fixture (16 mm × 16 mm × 10 mm) made of an acrylic panel, as shown schematically in our previous work,29) and EIS measurements were performed using a frequency-response analyzer (SI-1260, Solatron, England) following the same procedure mentioned therein.29) The total handling time before the EIS measurement was ~ 5 min. The impedance spectra were collected as a function of frequency at 10 points per decade in a logarithmic manner between 106-0.1 Hz using Z60 software.
3. Results and Discussion
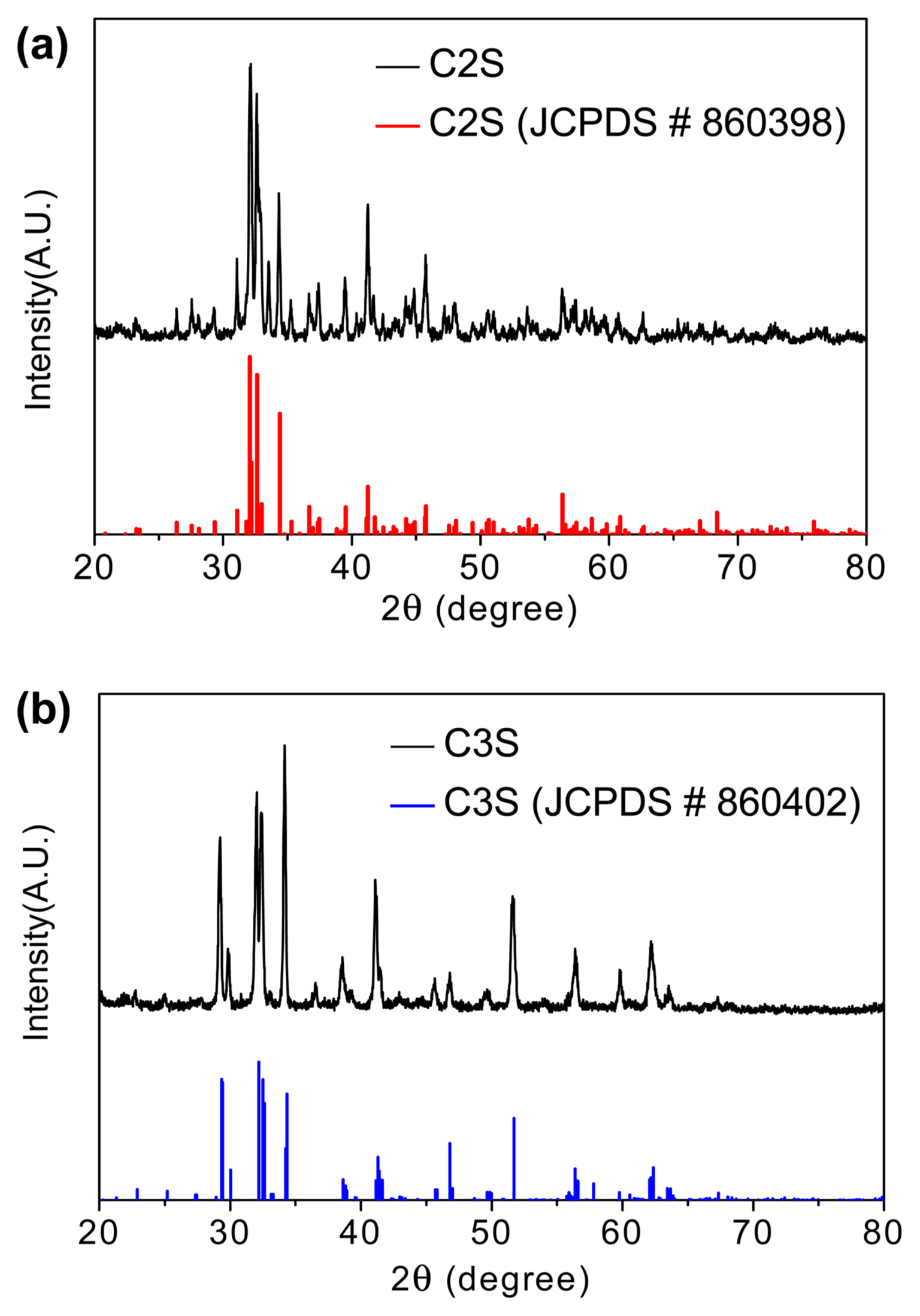
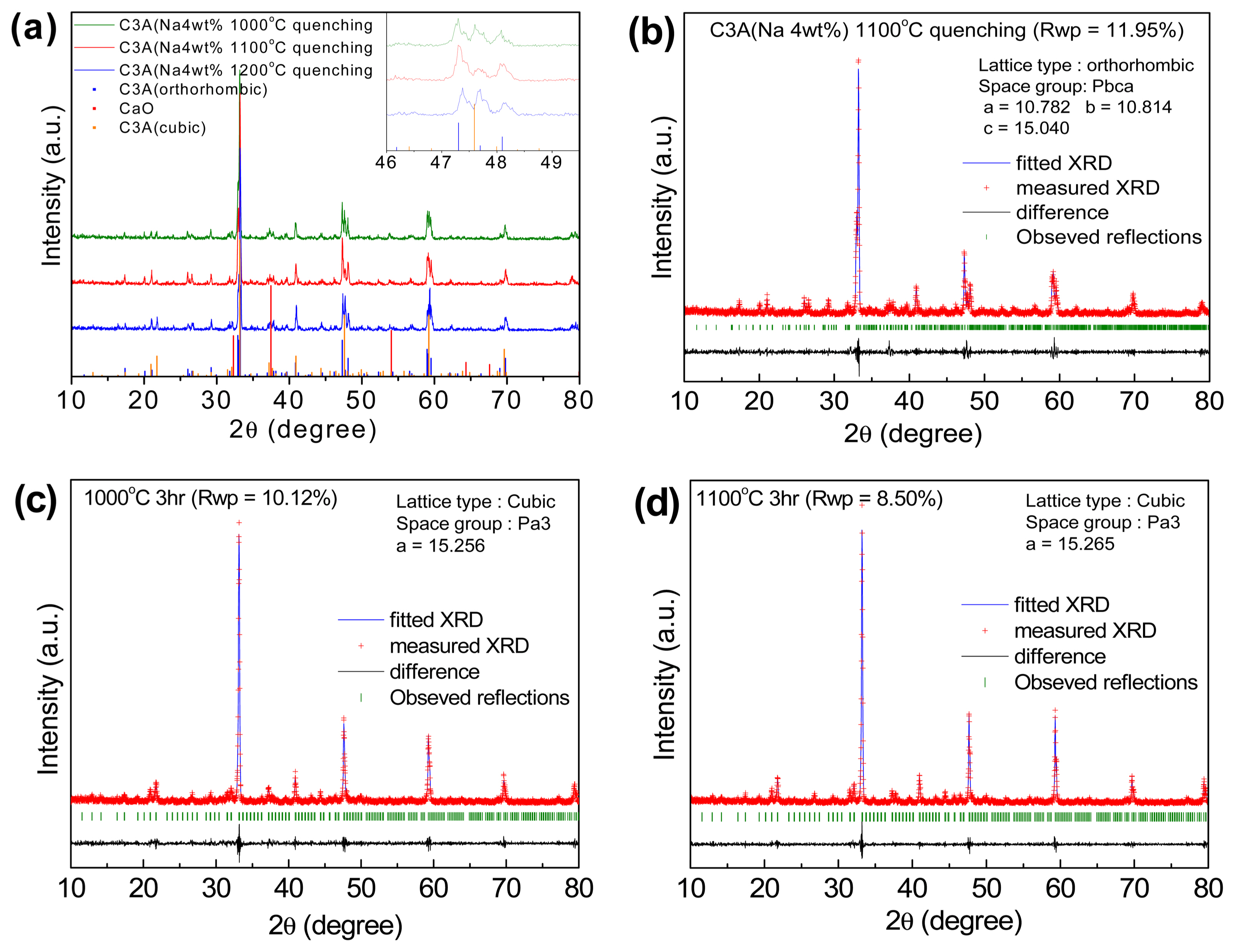
Figure 2 shows the XRD spectra of the C2S and C3S powders synthesized by solid-state reactions. The diffraction peaks in the C2S spectrum belong to β-C2S (JCPDS # 860398), whereas those in the C3S spectrum belong to monoclinic C3S (JCPDS # 860402), which indicate that these powders have high purity. Fig. 3(a) shows the XRD spectra of C3A synthesized by the Pechini method with sodium nitrite as one of the raw materials and quenched at different temperatures after sintering. Sodium nitrite was used to prepare Na+-substituted calcium aluminate.37) In the XRD spectrum of the 1000°C quenched sample, many peaks corresponding to the CaO phase and cubic C3A phase are prominently visible. The intensities of these peaks reduced upon quenching the sample at 1100°C, and the C3A sample mainly contains the orthorhombic C3A phase. However, for the 1200°C quenched sample, diffraction peaks corresponding to the cubic C3A phase again appeared. The lattice parameters of the 1100°C quenched sample calculated by Rietveld analysis considering the orthorhombic lattice (space group: Pbca) are a = 10.782 Å, b = 10.814 Å, and c = 15.040 Å. On the other hand, the XRD spectra of C3A prepared by the Pechini method without using sodium nitrite as one of the raw materials and quenched at 1000 and 1100°C after sintering are shown in Fig. 3(c) and 3(d). Rietveld analysis shows that these samples have a cubic lattice (space group: Pa3) with lattice parameters a = 15.256 Å and a = 15.265 Å, respectively. Although orthorhombic C3A is more reactive than cubic C3A38) during the hydration process, only the sample rich in orthorhombic C3A and quenched at 1100°C was used for the hydration studies.
The hydration process of the individual constituents, C3A, C2S, and C3S, was studied by EIS measurements. Impedance measurement allows the evaluation of the changes in ionic concentration of the pore solution as a result of microstructural development during the hydration process. The chemical reactions taking place during the hydration of the individual constituents, C3A, C2S, and C3S, can be represented as follows,39) where CH represents calcium hydroxide (Ca(OH)2):
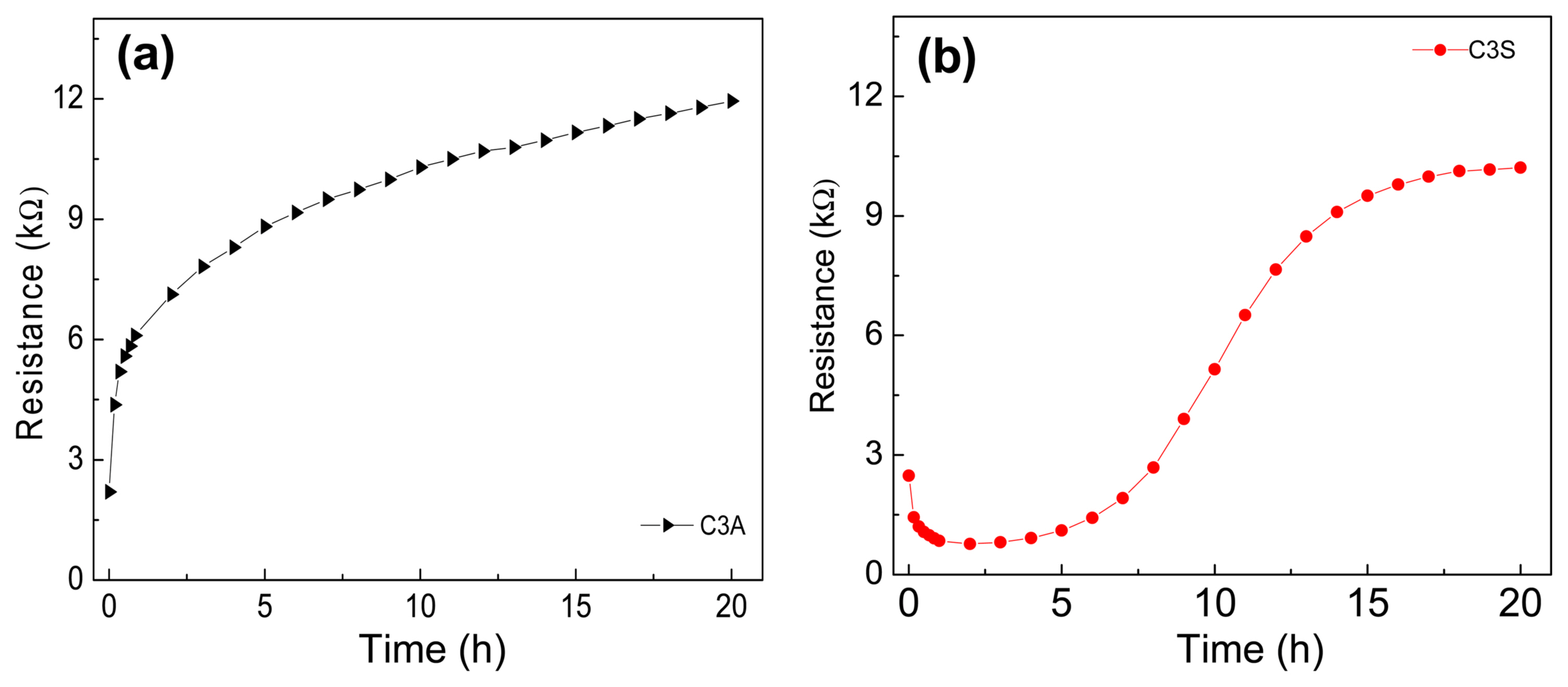
Considering the low-frequency intercept of the semi-circular part of the EIS data in the Nyquist plot during the hydration of C3A40) as a measure of the resistance of the C3A-water system, the resistances are plotted in Fig. 4(a). As can be seen, the resistance changes very rapidly in the initial 1-2 h and then gradually in the remaining period. It has been reported that C3A reacts almost instantaneously with water and is accompanied by the release of a large amount of heat and the formation of metastable hydrated products such as (Ca2[Al(OH)5]2·3H2O, 2[CaAl(OH)7·3H2O, and 4CaO·Al2O3·19H2O.41) In the present study, C3A shows a similar behavior.
Previously, the hydration of C3S and C2S has been studied by different analytical techniques.42-47) In the clinker composition, although the amount of C3A is 5-10 wt%, it has a strong influence on the setting and hardening times of cement because of its high reactivity with water.39,48) The values of resistances calculated from the variations in the impedance spectra during the hydration of C3S are plotted in Fig. 4(b). As can be seen, initially, the resistance decreases very rapidly in the initial 10-15 min and then gradually for the next 2 h due to the hydrolysis of C3S and the resultant release of calcium ions and hydroxide ion from the surface of the C3S grains.39) Once the dormant phase sets in and the concrete is in a plastic state, the resistance remains nearly unchanged for about the next 2 h. When the setting of concrete starts, the resistance increases again, first gradually and then rapidly. As early hardening begins, calcium and hydroxide ions start crystallizing, and hydration products are continuously formed. The hydration products form a thick coating around the anhydrous C3S grains and force the water to diffuse through the coating to react with the unreacted C3S grains.49) Under this diffusion-controlled phase, the resistance increases steadily. Fig. 5 shows the variations in the Nyquist plot during the hydration of C2S. It has been reported that the hydration of C2S is much slower than C3S hydration, and as a result, the resistance decreases very slowly during the hydration of C2S. This is because of its lower reactivity compared with C3S, that is, although the same calcium and hydroxide ions are released, albeit in lesser in numbers, they did not cause a rapid decrease in resistance during the hydration of C2S. Due to its slower rate of hydration, C2S does not contribute to initial strength, and therefore, in the present work, we did not study the effect of C2S on the initial hydration of our experimental cement.
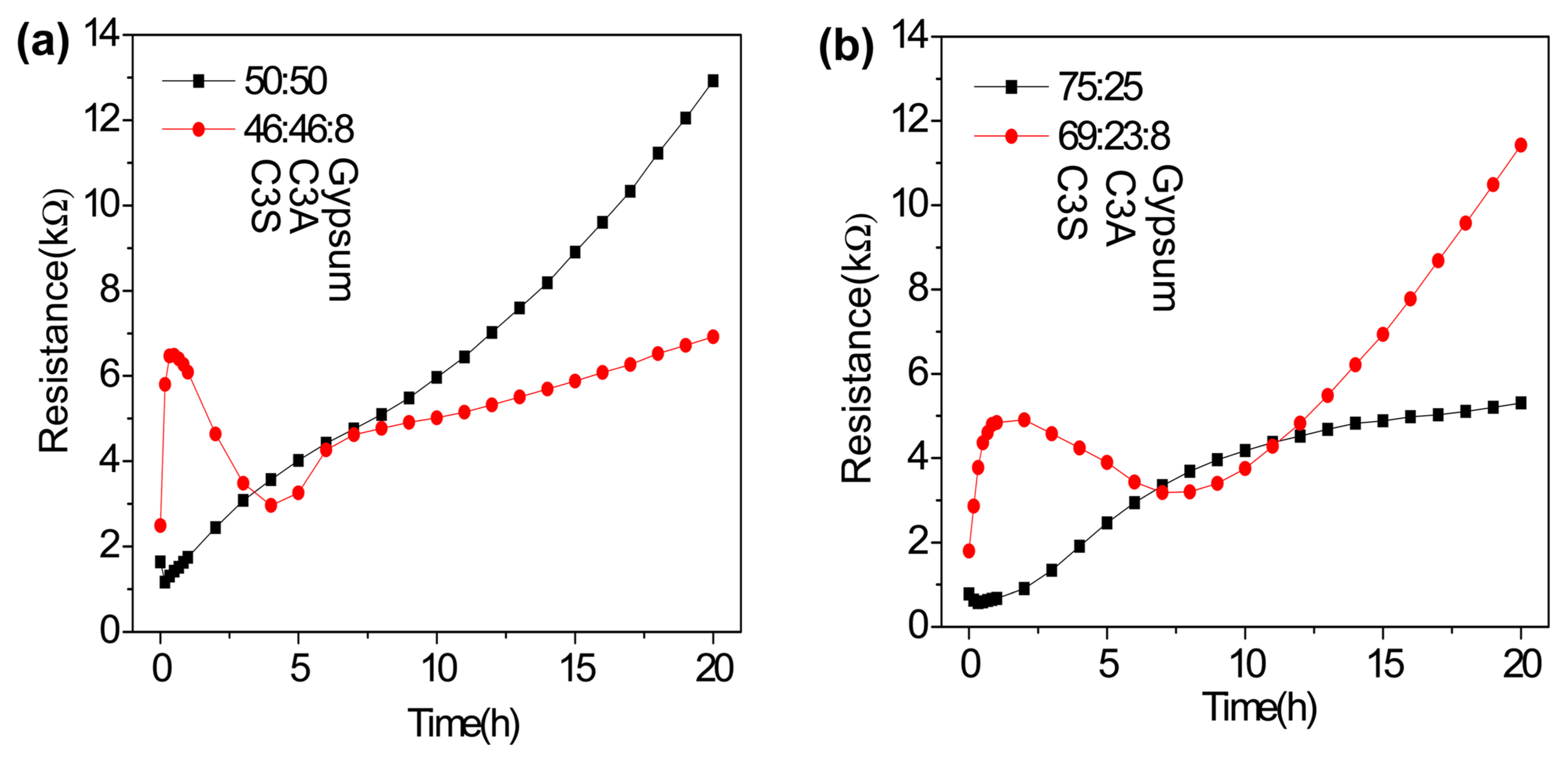
C3A undergoes fast hydration, and the addition of gypsum (calcium sulfate) has been reported to slow down the hydration reaction.38) In Portland cement, it is generally accepted that the effect of the addition of gypsum is mainly associated with its intensive reaction with the C3A phase. However, many studies have shown the indirect effect of gypsum on the hydration of silicate phases, with its addition resulting in the acceleration of C3S hydration and improvement in the strength of the C3S paste by entering C-S-H in some way.50-52) Therefore, we subsequently studied the hydration behaviors of C3A and C3S mixtures in the absence (50:50 and 25:75 wt% of C3A and C3S) and presence (46:46 and 23:69 wt% of C3A and C3S, and additional 8 wt% gypsum) of gypsum by EIS. The variations in resistance during the hydration of these mixtures are plotted in Fig. 6. In the absence of gypsum (Fig. 6(a)), the change in resistance of the C3A+C3S mixture follows an intermediate trend with a rapid initial decrease and a subsequent gradual increase in the resistance of the mixture. However, in the presence of gypsum (Fig. 6(b)), the change in the resistance of the C3A+C3S mixture is initially dominated by the change in the resistance of C3A, which is followed by a behavior intermediate to those of the two components. The change in the setting behavior of the mixture after ~ 10 h in the presence of gypsum is shown in Fig. 7; for the mixture having equal amounts of C3S and C3A, the resistance increases at a much slower rate during the subsequent setting process (Fig. 7(a)), while for the mixture having a higher amount of C3S, the resistance increases at a much faster rate during the subsequent setting process (Fig. 7(b)). This could be because with a lower C3A:C3S ratio, the amount of gypsum becomes higher than that required to suppress the hydration of C3A; therefore, the presence of a high amount of gypsum accelerates the setting of cement by generating a clotting agent.53)
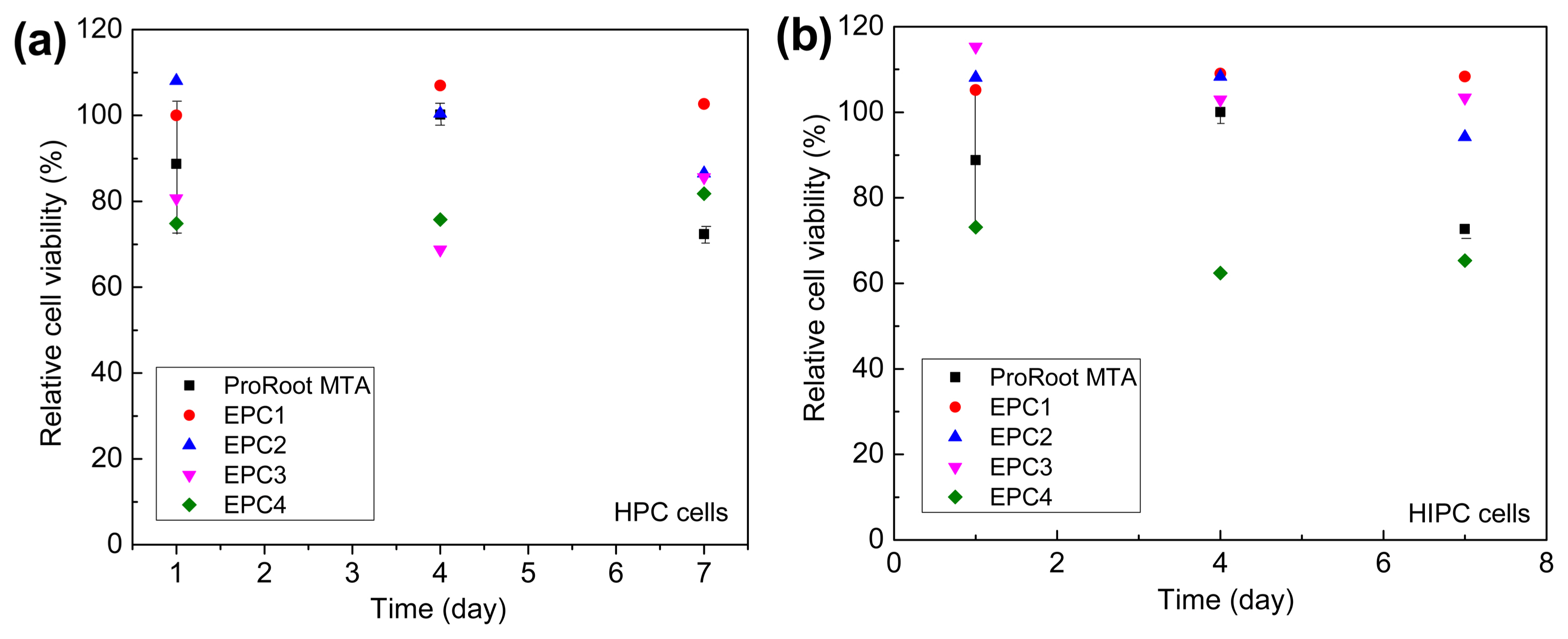
Furthermore, experimental dental cements with four different compositions (as mentioned in Table 2) were prepared using the lab-synthesized C3A, C3S, and C2S. However, since restorative materials contact or interact with body tissues and fluids, the biological compatibility of materials must be taken into account during material selection. Fig. 8 shows the cell viability of HPC and HIPC cells with cultured on the test materials. As can be seen, the cells cultured for 24 h on EPCs were viable and proliferated in direct contact with the cement surfaces. In addition, the EPCs and ProRoot MTA did not exhibit any significant cytotoxicity to HPC and HIPC cells for 7 days. Moreover, there is no significant difference in the cell viabilities of ProRoot MTA and the experimentally manufactured cements, which indicated that all the EPCs are sufficiently biocompatible with human dental pulp cells.
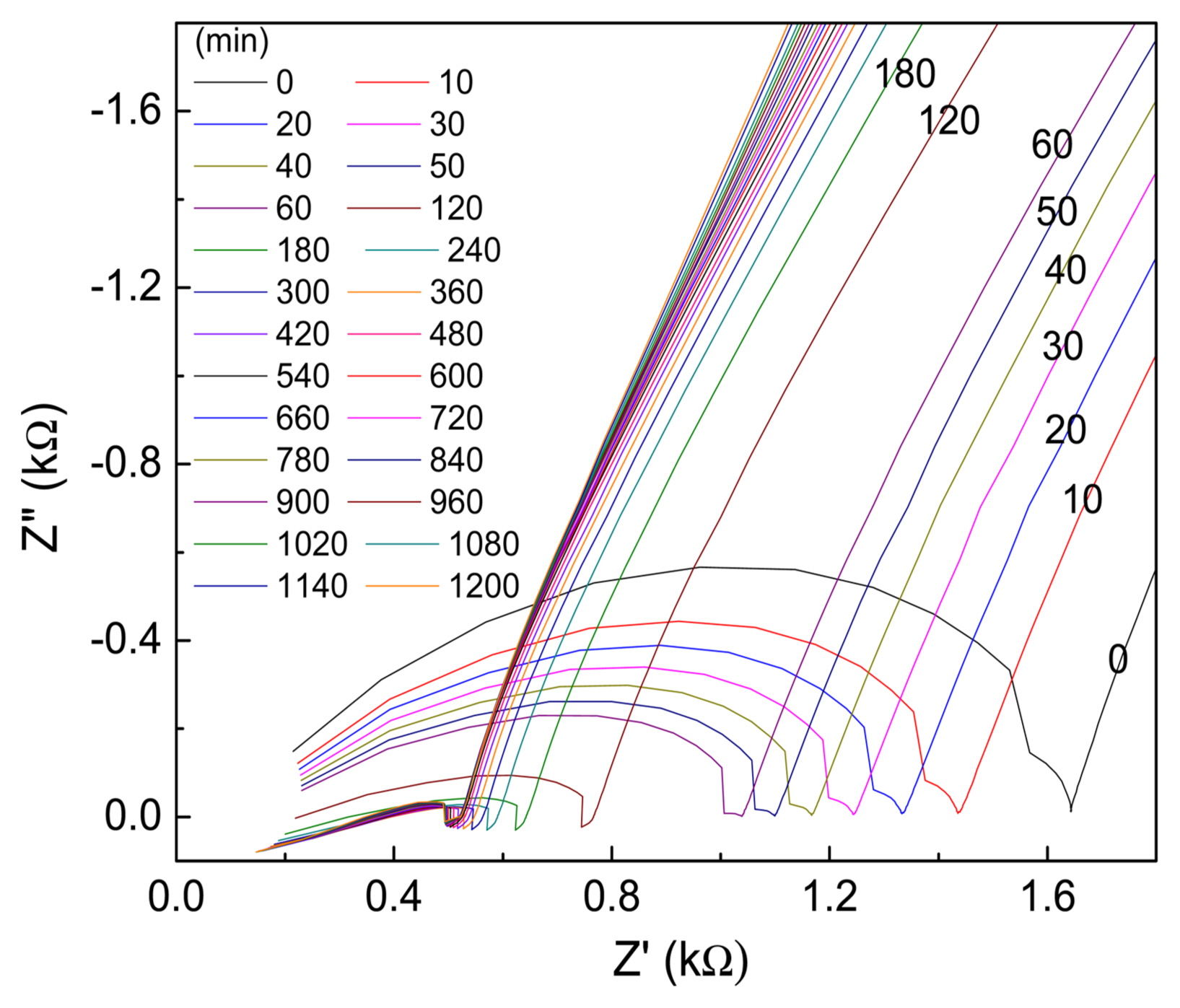
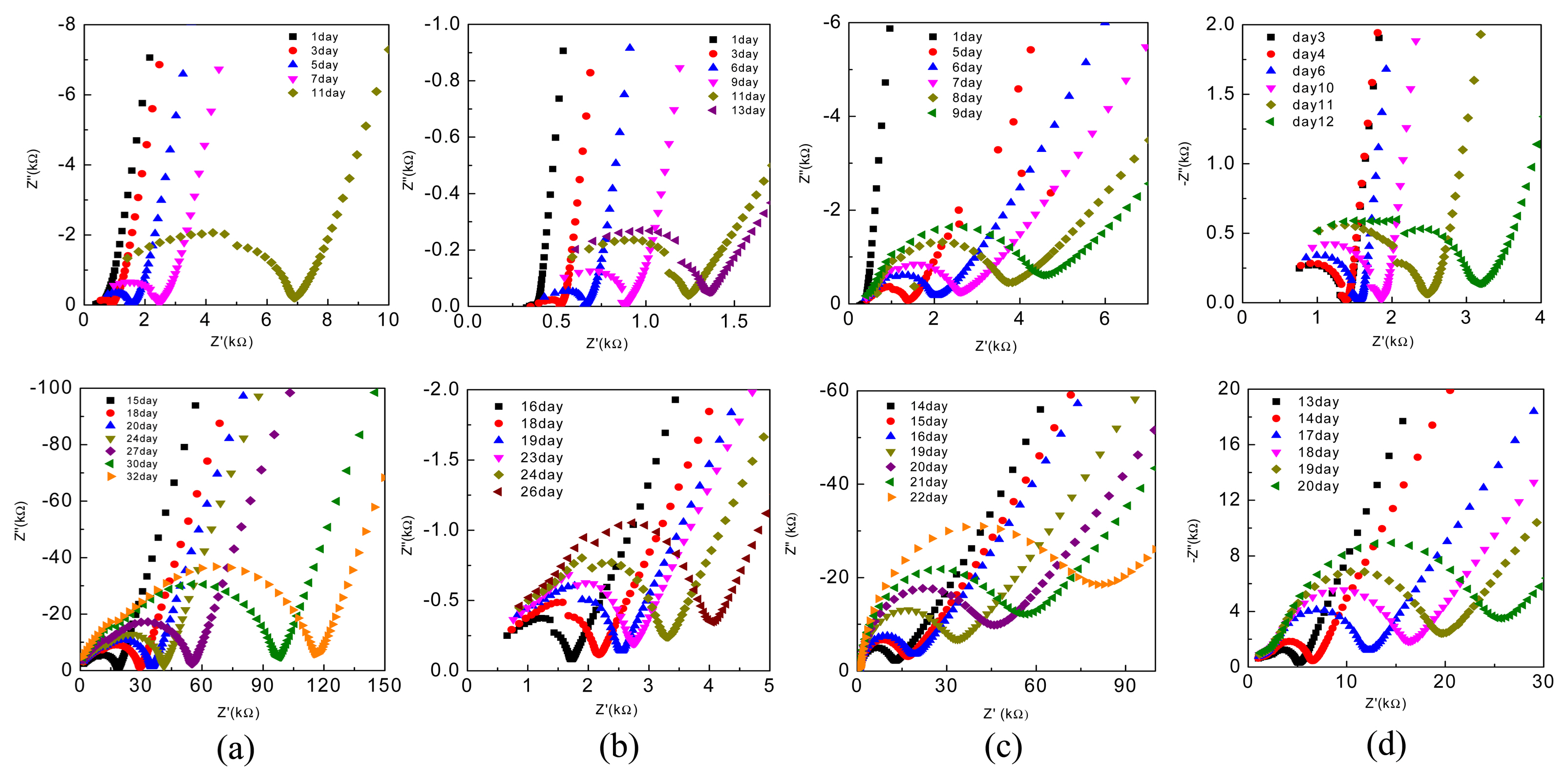
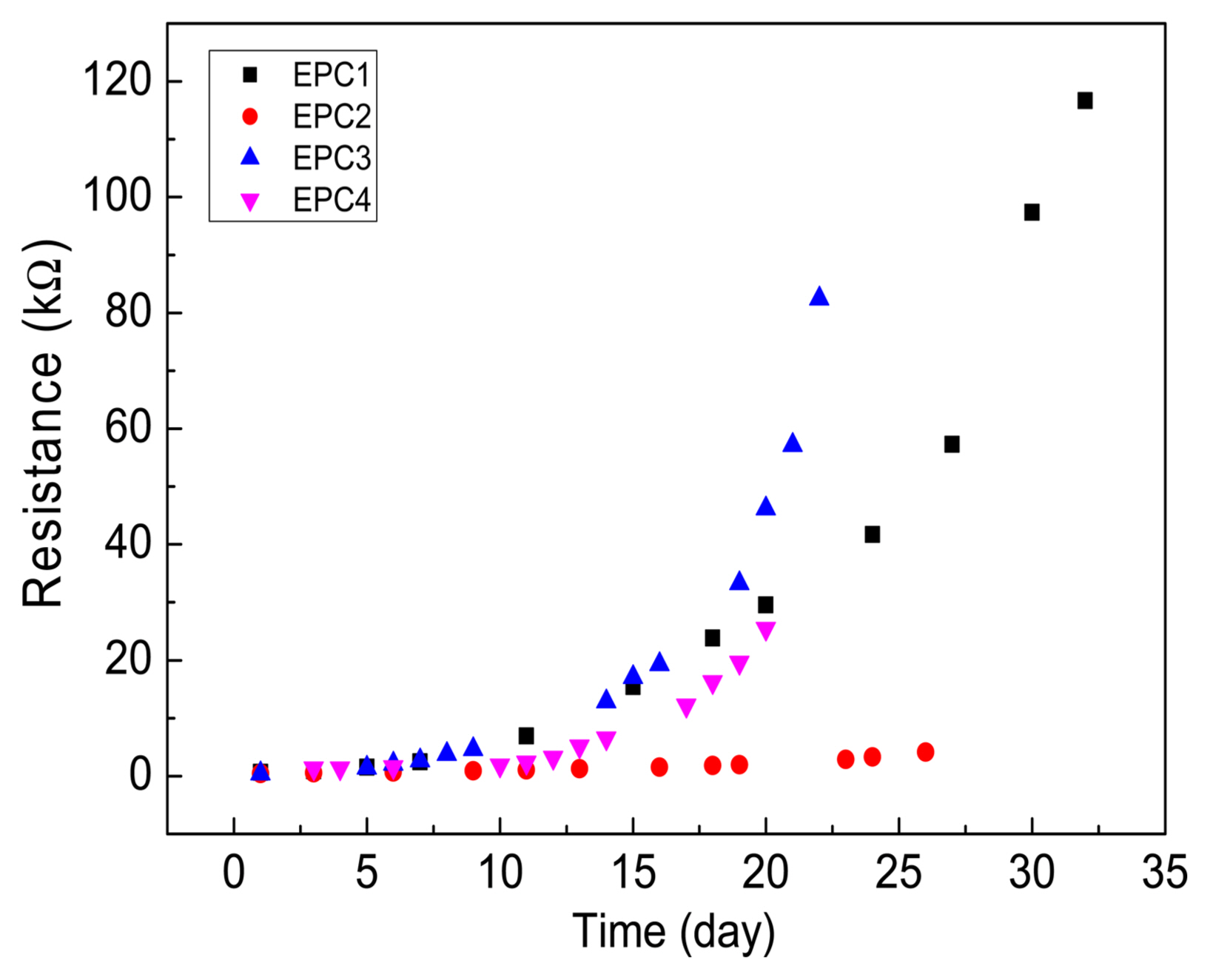
Finally, we studied the hydration process of the different EPCs by EIS and the changes in the Z′ vs. Z″ plots of the various EPCs are plotted in Fig. 9. During the setting of cement-based materials, the overall combination of solid network structure and composition of the ion-conducting liquid phase determines the electrical conductivity of the system.29) In addition, the continuously increasing solid phase penetrates the liquid phase due to the formation of silicate and hydrate products during the setting process, thus decreasing the porous ion-conducting pathways, which results in a continuous increase in the system’s resistance; this is indicated by the increase in the size of the high-frequency semicircles of the CPCs with setting time (Fig. 9). The changes in resistances during the hydration of the EPC compositions are plotted in Fig. 10. As can be seen, without gypsum (i.e., for EPC1 and EPC3), the resistance of EPC3 increases at a faster rate and reaches > 80 Ω in 22 days; however, during the same period, the resistance of EPC1 is < 40 Ω. On the other hand, for EPC2 and EPC4, the resistance increases at a much slower rate compared with EPC1 and EPC3, which indicates that the addition of gypsum retards the hydration process of EPCs. Among the EPCs with gypsum (i.e., EPC2 and EPC4), the resistance of EPC2 increases at a slower rate and is nearly one order of magnitude lower than that of EPC4 in 20 days.
4. Conclusions
In this work, three major clinker constituents of Portland cement—C3S, C2S, and C3A—were synthesized in the laboratory and their phase compositions were characterized by XRD analysis. C3A was synthesized by the Pechini method, whereas C3S and C2S were synthesized by solid-state reactions. The hydration process of the individual constituents and their combinations was investigated by EIS and the effect of the addition of gypsum on their hydration processes was examined. It was observed that the setting behavior of the mixture varied after ~ 10 h in the presence of gypsum; for the mixture having equal amounts of C3S and C3A, the resistance increased at a much slower rate during the subsequent setting process, whereas for the mixture with a higher amount of C3S, the resistance increased at a much faster rate during the subsequent setting process. This could be because with a lower C3A:C3S ratio, the amount of gypsum becomes higher than that required to suppress the hydration of C3A; therefore, the presence of a high amount of gypsum accelerates the setting of cement. Four different EPCs were prepared using the lab-synthesized C3A, C3S, and C2S, and their hydration processes were examined by EIS. In the XTT assay, the EPCs did not exhibit any significant cytotoxicity to HPC and HIPC cells for 7 days, and all the EPCs showed biocompatibility equivalent to that of the commercial cement, ProRoot MTA. This indicates that all the EPCs are potential substitutes for commercial dental cement.