Na-Ion Anode Based on Na(Li,Ti)O2 System: Effects of Mg Addition
Article information
Abstract
This study involves enhancing the performance of the Na(Li,Ti)O2 system as an Na-ion battery anode with the addition of Mg, which partially replaces Li ions. We perform both computational and experimental approaches to achieve a higher reversible capacity and a faster transport of Na ions for the devised system. Computational results indicate that the Na(Li,Mg,Ti)O2 system can provide a lower-barrier path for Na-ion diffusion than can a system without the addition of Mg. Experimentally, we synthesize various Naz(Liy,Mgx,Ti)O2 systems and evaluate their electrochemical characteristics. In agreement with the theoretical study, Mg addition to such systems improves general cell performance. For example, the prepared Na0.646(Li0.207Mg0.013Ti0.78)O2 system displays an increase in reversible capacity of 8.5% and in rate performance of 13.5%, compared to those characteristics of a system without the addition of Mg. Computational results indicate that these improvements can be attributed to the slight widening of the Na-O6 layer in the presence of Mg in the (Li,Ti)O6 layer.
1. Introduction
Green electricity from solar and wind energy requires large-scale energy storage, and electrochemical systems such as Li-ion batteries appear to be an effective and promising solution.1–3) Because Li-ion batteries can provide high efficiency as well as excellent energy density, it is common practice these days to use such batteries as rechargeable energy sources in small electronic devices, motors, and electric automobiles.4,5) The relatively high price of Li-ion batteries, however, is a critical drawback, especially when we consider it for application to large-scale energy storage.6,7)
Na-ion batteries have been considered as alternatives because of their low price and the abundance of Na resources; however, the energy density and efficiency of Na-ion batteries are lower than those of Li-ion batteries. It should be noted that the price of raw materials takes a higher priority when large-scale energy storage is concerned.8–10) There have been reported several non-metallic anodes for Na-ion batteries operating at room temperature such as hard carbon,11) Na2Ti3O7,12) P2-Na0.66(Li0.22Ti0.78)O2,13) etc. Here, P2 represents a hexagonal layered structure having transition metal (TM) layers, O layers in the sequence of ABBA, and Na layers in trigonal–prisms of 6 oxygen atoms (see Fig. 1; more details will follow in later section).
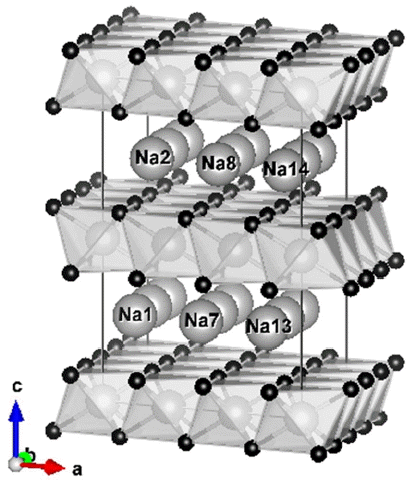
Supercell model of P2 phase, Na18(Li6Ti12)O36.
Hard carbon has high reversible capacity (240 mAh·g−1) and excellent cycle characteristics, but, because most of its discharge takes place at low voltages of 0.0 ~ 0.1 V (vs. Na+/ Na), it has the problem of dendrite growth by Na precipitation, and thus causes safety problems such as thermal runaway. 11) Na2Ti3O7, on the other hand, has a problem of shortened lifetime due to the large volume change associated with the insertion and extraction of Na ions.12)
Study of the Na0.66(Li0.22Ti0.78)O2 system has showed very promising aspects of the layered P2 system that can solve the above-mentioned drawbacks: its average redox voltage is 0.75 V, eliminating the possibility of Na precipitation; its volume change by insertion and extraction of Na ion is as low as 0.77%.13) In addition, the study estimated that the apparent Na+ diffusion coefficient is about 1 x 10−10 cm−2s−1 which is high enough to give fast charge and discharge capabilities. This fast diffusion of Na ions is attributed to the unique structure of Na0.66(Li0.22Ti0.78)O2, in which Na ions occupy relatively spacious prismatic sites made of 6 oxygen atoms between (Li,Ti)O6 octahedral layers.14–16) However, because of the limited empty sites for the reversible insertion and extraction of Na ions in the Na layers, the theoretical capacity of this material is limited to only 106 mAh·g−1.
In this study, we attempt to increase the reversible capacity of P2-Na0.66(Li0.22Ti0.78)O2 by partially substituting Li with Mg; we also attempt to reduce the Na fraction to less than 0.66 (this corresponds to increasing the ratio of Na sites for discharge to a value higher than 0.34), which has never been reported. To evaluate the possibility of effecting such a change, we first take a computational approach based on DFT, and carry out experiments to confirm the results by synthesizing the phase and analyzing it in terms of reversible capacity and rate capability. Based on the results, we evaluate the effect of Mg addition and identify its role.
2. Experimental Procedure
2.1. Computational Study
We first performed DFT calculations on the Nax(Li,Mg, Ti)O2 systems to evaluate their transport properties and to provide theoretical support for the experiments. Calculations were carried out within the framework of density functional theory (DFT)17) using the Vienna Ab-initio Simulation Package (VASP)18) and MedeA-VASP.19) We relaxed the electrons by projector augmented wave (PAW) potentials20) with the exchange-correlation energy of the Perdew–Burke–Ernzerhof (PBE) parameterization21) within the generalized gradient approximation (GGA).22,23) We adopted Li(1s,2s,2p), Na(2p, 3s), Mg(2p, 3s), Ti(3d, 4s), and O(2s, 2p) orbital as valence states.
We used an energy cutoff of 500 eV and the gamma-centered scheme to generate a 2 × 2 × 2 k-point mesh (k-spacing = 0.5/Å). We confirmed the total energy convergence of less than 10−5 eV per atom and optimized the geometric structure using the conjugate gradient method until all forces fell below 0.02 eVÅ−1. We did not consider spin polarization, dipole correction, or strong-correlation, because these factors do not affect the calculations of the energy differences in this case.
We used Bader analysis to calculate the electron transfer between atoms with the MedeA-VASP program.19) For the barrier energy determinations, we employed the nudged elastic band (NEB) method using the VTST script24) with 6 images including the initial and final configurations within a force convergence of less than 0.05 eVÅ−1. In all systems in this study, the hexagonal geometry of the P2 phase was maintained. Illustrations related to these computational models are drawn using VESTA.25)
Referring to previous studies,15,16) we selected a reference model for this study as shown in Fig. 1. The structure belongs to the space group of P63/mmc with hexagonal crystal system, and is a 3 × 3 × 2 supercell of Na(Li0.33Ti0.67)O2 in which each octahedron in the transition metal (TM) layer consists of Li or Ti atoms (light grey balls) at the center surrounded by 6 O atoms (small black balls). It should be noted that Na ions (big white balls) move along the inner-layer path which makes 2D diffusion possible. Based on this reference system, computational models were generated as shown in Table 1 by adding divalent Mg atoms to Li sites and/or by removing Na ions to create certain degrees of discharge.

Calculated Valence Charges and Charges Transferred Relative to Other Atoms by Bader Analysis
Table 2 provides a summary of the Bader analysis which indicates the charge transferred for each constituting atom. It should be noted that all the metals transfer electrons to O atoms and maintain local electroneutrality. Table 2 shows the four systems modeled for calculation; systems are 3 × 3 × 2 supercells of P2 phase with and without Mg in the TM layer. Each model corresponds to primitive P2 phase as follows: Li6Ti12O36Na17≡Na[Li0.33Ti0.67]O2, Li5Mg1Ti12O36Na17 ≡ Na0.94[Li0.278Mg0.055Ti0.67]O2, Li6Ti12O36Na14≡Na0.78[Li0.33Ti0.67]O2, and Li5Mg1Ti12O36Na14≡Na0.78[Li0.278Mg0.055Ti0.67]O2. It should be noted that the models with 17 and 14 Na ions correspond to 82% and 35% of discharge, respectively.

Summary of Calculated Results for Four Computational Models of P2 Phase
The data conclusively confirm that the introduction of Mg ions into the TM layer of the P2 phase increases the lattice vector of the supercell along the c-axis, the average length of the Na-O bonds, and the average volume of the Na-O6 prisms. These changes thus lead to ease of transport for Na ions, as indicated by the reduced barrier energies for the Mg-incorporated systems. It should be noted that this tendency is consistent for both high- and low-discharged cases, and also that Na ion transport becomes more difficult as the system is further discharged due to Na–Na repulsion.
Figure 2 shows, as examples, trajectories of the Na#14 ion in Li6Ti12O36Na14 and Li5Mg1Ti12O36Na14 systems during NEB simulations; Fig. 3 provides curves of the energy barriers experienced by Na#14 ion when it moves from an initial site (marked as Na14) to an empty Na site (path length ≈ 3.1 Å).
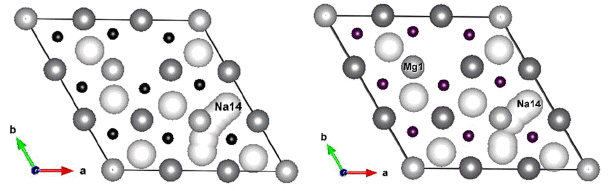
Trajectories of Na#14 ion in Li6Ti12O36Na14 (left) and Li5Mg1Ti12O36Na14 (right) systems during NEB simulations.
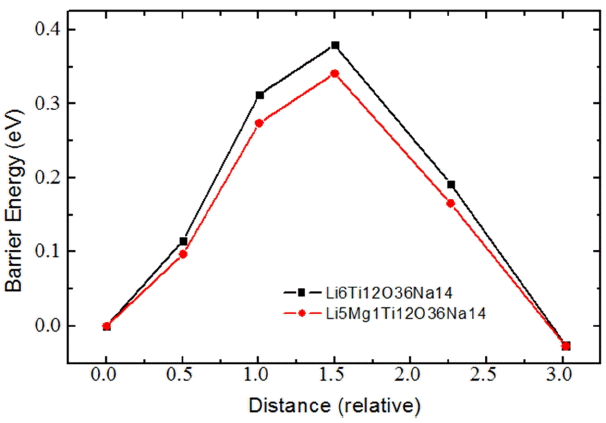
Curves of energy barriers experienced by Na#14 ion when it moves from an initial site (marked as Na14) to an empty Na site (path length ≈ 3.1 Å) in Li6− Ti12O36Na14 and Li5Mg1Ti12O36Na14.
2.2 Experimental
Raw materials for this study include Na2CO3 (Dae Jung Chem. ≥ 99.0%), Li2CO3 (Sigma Aldrich ≥ 99.0%), anatase TiO2 (Sigma Aldrich ≥ 99.8%), and MgCO3 (High Purity Chem. ≥ 99.9%). Table 3 shows the compositions considered and their theoretical capacities according to the various amounts of Li substituted with Mg. Each composition was prepared by mixing weighted raw materials with 250 ml of ethanol in a planetary mill (Fritsch, Pulverisette 5) with zirconia container and balls. The prepared slurries were dried in an evaporator, pressed into pellets, and heat-treated at 800, 850, 900, and 950°C for 20 h at heating rate of 5°C/min.

Compositions of P2 Phases Prepared for Anode and their Theoretical Capacity
The heat-treated samples were analyzed by X-Ray Diffraction (Rigaku, D/MAX-2500V) at 40 kV, 100 mA, 5°~90°, step interval of 0.05°, and scan speed of 4°/min; phases were identified with JCPDS No. 52-0689 as the reference for the P2-Na0.66(Li0.22Ti0.78)O2 phase. Microstructure was observed by scanning electron microscope (FEI, NOVA Nano-SEM200).
Electrochemical cells were prepared with anodes of the Nax(Liy−zMgzTi1−y)O2 system, cathodes of Na metal, and separators of glass microfibers. The anode samples were fabricated by mixing the synthesized anode powder, a conductive additive (Denka Black), and a binder (Polyvinylidenefluoride; PVdF 5 wt%) at a 8:1:1 weight ratio. The mixture was further mixed with NMP (1-methyl-2-pyrrolinone) to make the sample into paste form. The paste was casted on aluminum foil, dried, pressed, and punched into discs (ϕ12 mm) for use as anodes. The electrolyte consisted of 1: 1: 1 mol% of EC (ethylene carbonate), DEC (diethyl carbonate), and PC (propylene carbonate), with the addition of 1M NaClO4. The electrochemical behavior of the prepared cells was characterized by static-current method in a WBCS3000 battery cycler (WonATech) at a 0.4~2.5 V range. The C-rate was varied from C/20 to 5C.
3. Results and Discussion
Figure 4 shows the XRD results for the Na0.66(Li0.22Ti0.78)O2 sample in terms of the heat-treatment temperature. They indicate that the P2 phase formed at all temperatures. It should be noted that the peaks from the basal, prismatic, and pyramidal planes of the P2 phase well developed up to 850°C. However, unintended peaks of LiTi2O4 and NaTi2O4 phases appeared at 800°C. At temperatures above 900°C, peaks other than those from the basal plane of P2 phase weakened rapidly, while peaks of the new Na2Ti3O7 phase started to appear.

XRD patterns of P2 phases at various heat-treatment temperatures.
Figure 5 shows SEM images of the P2 phase. Powders prepared at 800 and 850°C show small (< 5 μm) and rounded grains, while those treated at temperatures higher than 900°C have grains bigger than 10 μm, are angular in shape, and have layers, as marked with the arrows. This could explain why peaks other than those from the basal plane of P2 phase weakened rapidly at this temperature.

SEM images of Na0.66(Li0.22Ti0.78)O2 powders annealed for 20 h at (a) 800, (b) 850, (c) 900, and (d) 950°C.
Figure 6 shows the XRD results of the P2 phase in which part of the Li is replaced with Mg. These results indicate that the substitution takes place up to 0.013 mole of Mg, and can increase up to 0.055 mole of Mg with minor formation of Na2Ti3O7 phase. Further substitution resulted in the formation of more secondary phases such as Na0.9Mg0.45Ti1.55O4 and MgTi2O5. The limit of Mg substitution is supposed to be less than 0.026 mole (actually 0.013 mole) in our experiment (From now on, we represent the degree of Mg substitution as x in unit of moles).

Figure 7 shows the results of the cycle test with a C-rate of less than 0.2C; results are summarized in Table 4. The best charge capacity and efficiency were found to result from the P2 phase, in which 0.013 mole of Mg is incorporated into the system. In this case, the initial charge capacity and efficiency at 0.05C improve by 8.5 and 7%, respectively, compared to the sample without Mg substitution. On the other hand, further substitution rather decreased both the charge and the discharge capacity, as well as the coulombic efficiency. The unintended substance Na2Ti3O7 that appeared in the powders with 0.026 and 0.055 moles of Mg also is an active material. The average discharge and charge potentials of Na2Ti3O7 are 0.2 and 0.4 V, respectively.12) It was found that the Na2Ti3O7 phase, however, did not work as an active material because the cutoff voltage was 0.4–2.5 V in this study. This may be one of the reasons for the lower capacity and coulombic efficiency of the powders with more than 0.013 mole Mg.


Average Charge Capacity at Various C-rates and Coulombic Efficiency for the First Cycle of P2-Na0.66−x (Li0.22-xMgxTi0.78)O2
Figure 8 shows charge/discharge curves of the P2 phase with 0.013 mole of Mg and without Mg and shows that the P2 phase with 0.013 mole of Mg has a higher discharge potential and lower charge potential; these results are indicative of the lower overpotential during charge/discharge. This observation is in agreement with the lower barrier energy for Na diffusion, calculated by the DFT method for the P2 phase with Mg substitution.
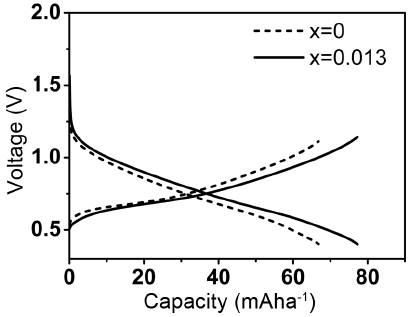
Charge/discharge curves of P2 phases with x = 0 and 0.013 at C/2.
Figure 9 shows the results of the charge/discharge capacity measured at a faster charge/discharge rate of up to 5C and 0.1C at the end. The P2 phase with Mg substitution maintains 77% of the initial capacity even at the faster charge/discharge rate of 5C, and fully recovers the initial capacity when the rate is reduced to 0.1C. This confirms that P2-Na0.66(Li0.22Ti0.78)O2 is a very stable structure that undergoes only negligible volume change.
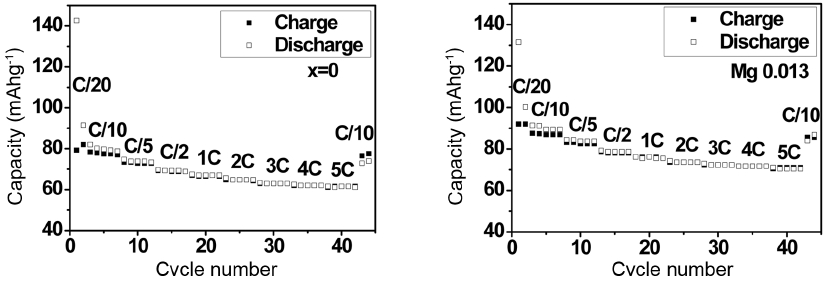
Rate capability of P2 phases with x = 0 and 0.013.
4. Conclusions
We prepare cells made of P2-type Na0.66(Li0.22Ti0.78)O2 phase as anode and introduce Mg2+ into the transition metal layer to take the part of Li+; this leads to a P2 phase with a Na ratio less than 0.66. The Mg-incorporated cells (0.013 ~ 0.026 mole of Mg) show a better electrochemical performance, as predicted by the first-principles calculation. Compared to the system without the addition of Mg, the prepared Na0.646(Li0.207Mg0.013Ti0.78)O2 system displays an increase in reversible capacity of 8.5% and an increase in rate performance of 13.5% at 1C. Computational results indicate that these improvements can be attributed to the widening of the Na layer by the presence of Mg in the (Li,Ti)O6 layer.
Acknowledgements
We would like to acknowledge the support of Materials Design (www.materialsdesign.com/medea) and Kyungwon Enc. (www.kwenc.kr), and to thank them for granting us the use of the MedeA-VASP program.